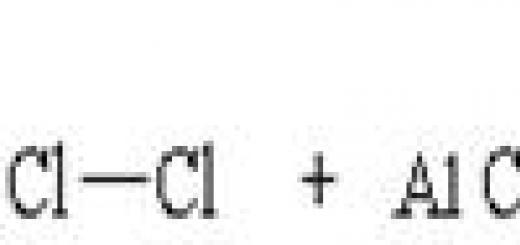Electronic theory of acids and bases put forward by Lewis in 1916 rejects the “cult of the proton.” It is based on consideration electronic structure of particles and does not consider the presence of hydrogen a mandatory sign of acid.
Acid– a particle with an unfilled outer electron shell, capable of accepting a pair of electrons ( acid= electron acceptor).
Base– particles with a free pair of electrons that can be donated to form a chemical bond ( base= electron donor).
TO acids according to Lewis: molecules formed by atoms with an empty eight-electron shell ( B.F.3 , SO3 ); complexing cations ( Fe3+ , Co2+ , Ag+ , etc.); halides with unsaturated bonds ( TiCl4 , SnCl4 ); molecules with polarized double bonds ( CO2 , SO2 ) and etc.
TO reasons According to Lewis, they include: molecules containing free electron pairs ( N.H.3 , H2 O);anions ( WITHl– , F– ); organic compounds with double and triple bonds (acetone CH3COCH3); aromatic compounds (aniline C6H5N.H.2 , phenol C6H5OH).ProtonH+ in Lewis theory it is an acid, (electron acceptor), hydroxide ionOH– – base (electron donor): HO–(↓) + H+ ↔ HO(↓)H.
The interaction between an acid and a base involves the formation of a chemical donor-acceptor bond between reacting particles. The reaction between an acid and a base in general: B(↓)base +Aacid ↔ D(↓) A.
Disadvantages of Lewis's theory To classify a substance as an acid or base, the mechanism of its formation is used, which puts classification for acids and bases in close dependence from views on the nature of chemical bonds. Lewis's theory has no quantitative criterion assessment of the strength of acids and bases, which could serve as a basis for analytical calculations of acid-base equilibria.
General theory of Usanovich. The most general theory of acids and bases was formulated by Usanovich in 1938.
The theory is based on the idea: “Every acid-base interaction is a salt formation reaction.”
Acid according to Usanovich, this is a particle that can remove cations, including a proton, or attach anions, including an electron. BaseBy Usanovich is a particle that can attach a proton and other cations or donate an electron and other anions. According to Usanovich's theory acid-base interactions All redox reactions also apply. Usanovich's theory is canceled one of the fundamental principles of classical chemistry is ideas about the classes of acids and bases.According to Usanovich, acids And grounds- This not classes connections; acidity And basicity- This functions substances. Whether a substance is an acid or a base depends on the partner in the reaction.
Disadvantages of Usanovich's general theory: too general nature of the theory; lack of clarity in the formulation of concepts " acid" And " base";does not include nonionic acid-base transformations; does not allow quantitative predictions to be made.
author unknownAccording to Lewis, the acidic and basic properties of organic compounds are assessed by their ability to accept or provide an electron pair and subsequently form a bond. An atom that accepts an electron pair is an electron acceptor, and a compound containing such an atom should be classified as an acid. The atom that provides an electron pair is an electron donor, and the compound containing such an atom is a base.
Compared to Brønsted's proton theory, Lewis's theory is more general and covers a wider range of compounds. Taking into account the energy characteristics of the orbitals involved in acid-base interactions, a Lewis acid is a molecule with a low-energy free molecular orbital, and a Lewis base is a molecule that provides a high-energy filled molecular orbital for intermolecular interactions. Specifically, Lewis acids can be an atom, molecule or cation: a proton, halides of elements of the second and third groups of the Periodic Table, halides of transition metals - BF3, ZnCl2, AlCl3, FeCl3, FeBr3, TiCl4, SnCl4, SbCl5, metal cations, sulfuric anhydride - SO3, carbocation. Lewis bases include amines (RNH2, R2NH, R3N), alcohols ROH, ethers ROR, thiols RSH, thioethers RSR, anions, compounds containing p-bonds (including aromatic and heterocyclic compounds), especially if their donating ability is enhanced electron-donating substituents.
Now we will try to compare two approaches (Brønsted and Lewis) to the determination of acids and bases. As can be seen from the definitions, Lewis bases are identical to Brønsted bases: both are donors of a pair of electrons. The only difference is where this electron pair is spent. Brønsted bases provide it for bonding with a proton and are therefore a special case of Lewis bases, which provide an electron pair to any particle with a vacant orbital. More significant differences are noted in the interpretation of acids. Brønsted's theory covers only protic acids, while Lewis acids are any compounds with a vacant orbital. Protic acids are considered in Lewis theory not as acids, but as products of proton neutralization by bases. For example, sulfuric acid is the product of neutralization of the acid H+ with a base, hydrochloric acid is the product of neutralization of H+ with the base Cl-.
When Lewis acids and bases interact, donor-acceptor (acid-base) complexes of various natures are formed. Below are examples of such interactions.
Organic chemistry is rich in examples of such interactions, in which a covalent bond is formed as a result of the interaction of a particle having a filled orbital with a particle having a vacant orbital. These processes can be thought of as Lewis acid-base reactions. The wider coverage of specific objects, characteristic of the Lewis theory, and more significant differences in the nature of the compounds lead to the fact that the Lewis series of relative strengths of acids and bases is not as universal as for Bronsted acids and bases. For Lewis acids it is impossible to compile a table with strict quantitative characteristics of acidity, as was done for Bronsted acids (see Table 1). For them there is only a qualitative approximate sequence of acidity. Thus, for Lewis acids such as metal halides, the acidity decreases in the series: BX3 > AlX3 > FeX3 > SbX5 > SnX4 > ZnX2.
Summarizing the above, we note that at present there are two theories in assessing the acid-base properties of organic compounds. Can we say that one of them has significant advantages over the other? There can be no clear answer to such a question. Yes, Lewis's theory is more general and covers a wider range of specific objects. The Brønsted-Lowry theory is characterized by a more stringent account of the quantitative characteristics of acidity and basicity. Preference for one theory or another can be given only taking into account the specific content of the issue under discussion. If processes occurring with the participation of hydrogen-containing substances are discussed, in which proton transfer reactions play an important role and hydrogen bonds have a significant influence, apparently, in these cases, preference should be given to the Brønsted-Lowry theory. An important advantage of Lewis's theory is that any organic compound can be represented as an acid-base complex. When discussing heterolytic reactions in which Lewis acids participate as electrophilic reagents and Lewis bases as nucleophiles, preference should be given to the Lewis theory. Chemists have learned to skillfully use the advantages of each of these theories.
A Lewis acid is a molecule or ion that has vacant electron orbitals, as a result of which it is capable of accepting electron pairs. For example, hydrogen ions - protons, metal ions (Ag +, Fe 3+), oxides of some non-metals (SO 3, SiO 2), a number of salts (AlCl 3), substances like BF 3, Al 2 O 3. Lewis acids that do not contain hydrogen ions are called aprotic acids. Protic acids are considered as a special case of the class of acids.
A Lewis base is a molecule or ion capable of donating electron pairs: all anions, ammonia and amines, water, alcohols, halogens.
Examples of chemical reactions between Lewis acids and bases:
· AlCl 3 + Cl − → AlCl 4 −
· BF 3 + F − → BF 4 −
· PCl 5 + Cl − → PCl 6 − .
Ionic potential is the ratio of the electronic charge of an ion to its effective radius.
Expressed by the ratio Z/r, where Z is the charge, r - ion radius. Used to characterize the interaction of an ion in a crystal lattice or in solution
Hard bases include donor particles that have high electronegativity, low polarizability, and are difficult to oxidize. The compound holds its electrons tightly, its molecular orbital, a pair of electrons of which is transferred to the acceptor, has a low energy level Soft bases. These include donor particles with low electronegativity, high polarizability, and are quite easily oxidized. They weakly retain their valence electrons, their molecular orbitals, and have a high level of energy (the electrons are removed from the nucleus of the atom).
Hard acids. include Lewis acids, in which the acceptor atoms are small in size, have a large positive charge, high electronegativity and low polarizability. A molecular orbital has a low energy level. Soft acids. include Lewis acids containing large acceptor atoms with a small positive charge, low electronegativity and high polarizability. A molecular orbital has a high energy level. The essence of the HMCO principle is that hard acids react preferentially with hard bases, and soft acids with soft bases. higher reaction rates for the formation of more stable compounds
Ticket number 2 1. Halogens. Oxidation states. Disproportionation of halogens. Comparison of oxidizing capacity. Hydrogen halides and hydrohalic acids. Features of HF. Metal and non-metal halides, their interaction with water. Halogen oxides.
In the ground state, halogen atoms have the electronic configuration nsnр5. fluorine to a smaller radius, higher values of ionization energy and electronegativity. The electron affinity of fluorine is less than that of chlorine. fluorine oxidation states -1, 0.
Halogen compounds in positive oxidation states exhibit oxidizing properties.
Halogens are the most reactive nonmetals. Fluorine interacts with almost all simple substances, with the exception of light inert gases. From fluorine to iodine, the oxidizing ability decreases, and the reducing ability increases. Chlorine reacts with oxides of some metals: magnesium, aluminum, iron.
2MgO + 2C12 = 2MgCl2 + 02
Bromine is a strong oxidizing agent. In an aqueous environment, it oxidizes sulfur to sulfuric acid:
ZVg2 + S + 4Н20 = bНВg + H2S04
potassium manganate - to permanganate:
2K2Mp04 + Vg2 = 2KMp04 + 2KVg
The oxidizing properties of iodine are less pronounced than other halogens. Iodine is not able to oxidize not only oxygen, but also sulfur. Iodides have reducing properties. Under the influence of chlorine, bromine, hydrogen peroxide and nitric acid, it is oxidized in an aqueous medium to iodic acid H03:
3I2(tv) + 10HNO3(100%) = 6НIO3 + 10NO2 + 2Н20
Under standard conditions, hydrogen halides are colorless gases with a pungent odor. for HF, the values of the melting and boiling points. The abnormally high melting and boiling points of hydrogen fluoride are explained by increased intermolecular interaction due to the formation of hydrogen bonds between HF molecules. Solid hydrogen fluoride consists of
zigzag polymer chains. For HCI, HBr, HI, the formation of hydrogen bonds is not typical due to the lower electronegativity of the halogen atom. Aqueous solutions of HC1, HBr and HI behave like strong acids. hydrofluoric HF and hydrochloric acids HC1 do not interact with concentrated sulfuric acid, but HBr and HI are oxidized by it:
2HBr + H2S04(koh4.) = Br2t + S02 + 2H20
8HI + H2S04(koh4.) = 4I2 + H2S + 4H20
Alkali and alkaline earth metal halides are ionic substances. They are soluble in water and have high melting and boiling points.
Hypohalogenitic acids NHO are known only in dilute aqueous solutions.
Hypohalogenated acids are weak. acidic properties in the series HSIu-HBrO-Niu weaken, and basic properties increase. Hydrous acid is already an amphoteric compound.
Solutions of hypohalogenites have a highly alkaline reaction, and passing CO2 through them leads to the formation of acid:
NaCIO + Н20 + С02 = NaHC03 + НСУ
Hypohalogenated acids and their salts are strong oxidizing agents:
Among the oxoacids HXO2, chlorous acid HXO2 is known.
HClO2 is a medium strength acid.
HXO3 oxoacids are more stable than hypohalite acids. Hypochlorous HSO3 and bromic HSO3 acids were obtained in solutions with concentrations below 50%, and iodic HSO3 was isolated as an individual substance. Solutions of HClO3 and HBrO3 are obtained by the action of dilute H2S04 on solutions of the corresponding salts, for example:
Ba(ClO3)2 + H2S04 = 2HClO3 + BaS04
Iodonic acid is obtained by oxidation of iodine with fuming nitric acid.
acid, hydrogen peroxide solution:
I2 + 5Н202 = 2НI3 + 4Н20.
HXO3 are strong acids. In the series HClO3 - HBrO3 - HI3 there is a slight decrease in the strength of the acids.
Perchloric acid HC104. is released in the form of hydrates HC104*H20. Bromic acid HBrO4 is known only in solutions.
Liquid HF is composed of HF polymer chains.
The halogen-oxygen bond is fragile, which is caused by the strong mutual repulsion of atoms with high
Electronegativity. halogen oxides are unstable. Oxygen difluoride OF2 can be obtained
2F2 + 2NaOH =OF2 + 2NaF + H20
Oxygen difluoride is a strong oxidizing-fluorinating agent.
By passing an electric discharge through a cooled mixture of fluorine and oxygen, another fluoride, 02F2, can be obtained.
Chlorine oxide (I) C120 It is obtained
3HgO + 2С1 2 = Hg30 2 Cl 2 + Cl 2 O
The connection is extremely unstable.
2. Titanium, zirconium, hafnium. Comparison of redox properties. Interaction of metals with solutions of acids and alkalis. Difference between Ti compounds and Zr and Hf. Reactions of Ti 2+ and Ti 3+ compounds. E 4+ compounds: oxides, a- and b-forms of acids. Halides, their hydrolysis. Oxocation salts. Halide complexes.
Ionization noticeably decreases when moving from titanium to zirconium.
Only the first of the elements of the group, titanium, exhibits high chemical activity. Hafnium has lanthanide compression. The characteristic oxidation state is +4, most compounds are covalent. In the Ti - Zr-Hf series, the stability of compounds with the highest oxidation state increases. Thus, for titanium, the oxides TiO, Ti2O3, Ti02 and fluorides TiF2, TiF3, TiF4 are stable, and for zirconium and hafnium - only dioxides ZrO2, Hf02 and tetrafluorides ZrF4, HfF4. The tendency for titanium to exhibit low oxidation states +2, +3 is higher than that of its heavy analogues. Zirconium(III) and hafnium(III) compounds do not exist in aqueous solutions. oxidation states enhance basic and reducing properties
For titanium, the typical coordination number is 6 and, less commonly, 4; zirconium and hafnium 7 and 8.
The reaction with halogens begins with low heating; MX4 tetrahalides are always formed.
Unlike zirconium and hafnium, titanium reacts with hydrochloric and dilute sulfuric acids when heated
2Ti + 6HC1 = 2TiCl3 + ZN2T
Titanium also dissolves in concentrated hydrofluoric acid to form green solutions.
2Ti + 6HF = 2- + Ti2+ + ZN2T
Ti + 6HF + 02 = H2 + 2H20
Titanium dissolves extremely slowly in dilute and concentrated nitric acid, as well as in aqua regia - the reaction is prevented by the formation of a layer
Ti + 4H2S207 - Ti(S04)2 + 2S02T + 4H2S04
When heated, titanium powder slowly dissolves in concentrated solutions and melts of alkalis:
Ti + 2NaOH + H20 = Na2Ti03 + 2H2
Zirconium and especially hafnium are more resistant to oxidation by acids. do not react with any dilute acid except hydrofluoric acid. zirconium and hafnium react vigorously only with a mixture of nitric and hydrofluoric acids:
ЗМ + 4HN03+ 21HX = ЗН3[МХ7] + 4NO + 8Н20
The interaction of zirconium and hafnium with hydrofluoric acid and concentrated sulfuric acid occurs more slowly:
M + 7HF = H3 +2H2T
M + 5H2S04 = H2 + 2S02t + 4H20
Concentrated HN03 increases the corrosion resistance of metals. Zirconium and hafnium do not react with alkalis.
An idea of reaction mechanisms. Homo- and heterolytic bond breaking. Introduction to intermediate particles: radicals, carbocations, carbanions. Classification of reagents: radicals, nucleophiles, electrophiles.
A reaction mechanism is a detailed description of the process of converting reagents into products, including as complete a description as possible of the composition, structure, geometry, energy and other properties of intermediates, transition states and products.
Homolytic bond cleavage is a cleavage where each atom loses one electron. Characteristic of the exchange mechanism of covalent bond formation.
Heterolytic bond rupture is a rupture when positively and negatively charged particles are formed as a result, because both electrons from a common electron pair remain with one of the atoms. Characteristic of the donor-acceptor mechanism of covalent bond formation.
Carbocation is a particle, in a cat. A positive charge is concentrated on the carbon atom; the carbon atom has a vacant p-orbital. Carbocation is a strong Lewis acid and has electrophilic activity. Chemical saints:
· Interaction with nucleophiles.
· The ability for β-elimination - the removal of a proton to form a multiple bond.
· Rearrangement into a more stable carbocation - isomerization of a primary into a more stable secondary or tertiary carbocation.
A carbanion is an anion with an even number of electrons with a free electron pair on a tetravalent carbon atom. Carbanions include both anions with a negative charge localized on the carbon atom and anions with a delocalized negative charge, in which in at least one of the canonical structures the charge is localized on the carbon atom. Chem. saints:
· Interaction with electrophiles.
· Oxidation to radicals.
Free radicals are particles (usually unstable), containing one or more. unpaired electrons to the outer electron. shell. A radical can be formed as a result of the loss of one electron by a non-radical molecule or by the gain of one electron by a non-radical molecule.
Acids and Bases (Brønsted, Lewis)
Protolytic (proton) theory of acids and bases by Brønsted - Lowry (1923). According to this theory, acids are molecules or ions that can be proton donors in a given reaction, and bases are molecules or ions that add protons (acceptors). Acids and bases are collectively called protolytes.
The essence of acid-base interaction is transfer of a proton from an acid to a base. In this case, the acid, having transferred a proton to the base, itself becomes a base, because can reacquire a proton, and the base, forming a protonated species, becomes an acid. Thus, any acid-base interaction involves two pairs of acids and bases, called conjugate by Brønsted.
Lewis's electron theory. In the theory of Lewis (1923), based on electronic concepts, the concept of acid and base was further expanded. A Lewis acid is a molecule or ion that has vacant electron orbitals, as a result of which it is capable of accepting electron pairs. These are, for example, H ions - protons, metal ions (Ag+, Fe3+), oxides of some non-metals (for example, SO3, SiO2), a number of salts (AlCl3), as well as such things as BF3, Al2O3. Lewis acids that do not contain hydrogen ions are called aprotic acids. Protic acids are considered as a special case of the class of acids. A Lewis base is a molecule or ion capable of donating electron pairs: all anions, ammonia and amines, water, alcohols, halogens.
Ways to use alkanes
Alkanes are widely used in many areas of human activity. None of us can imagine life without natural gas, the basis of the cat. is methane. Carbon black (soot) is also produced from it. used in the production of tires and printing ink. Alkane compounds are used as refrigerants in home refrigerators. Acetylene, cat. obtained from methane, used for welding and cutting metals. Among the compounds of alkanes, halogen derivatives can be distinguished, such as chloroform and carbon tetrachloride, which are among the best solvents. Alkanes can be used as motor fuel (methane, propane, butane), cat. produces little environmental pollution. Vaseline oil (a mixture of liquid hydrocarbons with a number of carbon atoms up to 15) is a transparent, odorless and tasteless liquid, used in medicine, perfumery and cosmetics. Vaseline (a mixture of liquid and solid saturated hydrocarbons with a number of carbon atoms up to 25) is used for the preparation of ointments used in medicine. Paraffin (a mixture of solid alkanes C19-C35) is a white, odorless and tasteless solid mass (melting point = 50-70°C) - used for making candles, impregnating matches and wrapping paper, for thermal procedures in medicine, etc. .
Sp hybridization
Occurs when one s- and one p-orbital mix. Two equivalent sp-atomic orbitals are formed, located linearly at an angle of 180 degrees and directed in different directions from the nucleus of the central atom. The two remaining non-hybrid p-orbitals are located in mutually perpendicular planes and participate in the formation of π bonds or occupy lone pairs of electrons.
Sp2 hybridization
Occurs when one s- and two p-orbitals mix. Three hybrid orbitals are formed with axes located in the same plane and directed to the vertices of the triangle at an angle of 120 degrees. The non-hybrid p-atomic orbital is perpendicular to the plane and, as a rule, is involved in the formation of π bonds
26. Alkynes. Reduction of a triple bond to a double bond: catalytic hydrogenation and reduction with sodium in liquid ammonia, use in synthesis ( Z)- And ( E) - alkenes.
Triple bond represents one s-C-C bond and two p-bonds. When going from a double to a triple bond, the average p-bond energy decreases. This means that the triple bond is less stable than the double bond. Acetylene itself is an unstable compound and is capable of spontaneous explosive decomposition into elements. The acetylene molecule has a linear structure, which is due to the sp-state of the carbon atoms. The triple bond in alkynes is characterized by a higher polarizability than in alkenes R Cº C = 5.96; R C=C =4.17.
Catalytic hydrogenation method , along with other important processes of organic chemistry, is currently widely used. The introduction of hydrogenation into technology was a stimulus for the widespread development of fuel refining processes, syntheses from carbon oxides and numerous reduction reactions. The hydrogenation of alkynes occurs under approximately the same conditions and in the presence of the same catalysts as the hydrogenation of alkenes. The first stage of hydrogenation of acetylene to ethylene is more exothermic than the second, where ethylene is converted to ethane:
From these data it follows that the hydrogenation of alkynes, in principle, can be stopped at the stage of alkene formation. However, with most catalysts, alkynes are hydrogenated directly to alkanes:
Reduction of alkynes with sodium or lithium in liquid ammonia or in amines gives trance-alkenes:
Alkenes (olefins, ethylene hydrocarbons) - acyclic unsaturated hydrocarbons containing one double bond between carbon atoms, forming a homologous series with the general formula CnH2n. The carbon atoms at the double bond are in a state of sp² hybridization and have a bond angle of 120 °C. The simplest alkene is ethylene (C 2 H 4). According to nomenclature, the names of alkenes are formed from the names of the corresponding alkanes by replacing the suffix “-an” with “-ene”; The position of the double bond is indicated by an Arabic numeral. E-Isomers are geometric isomers in which the senior substituents on the carbon atoms of the double bond are located on opposite sides of the double bond. Z-Isomers are geometric isomers in which the senior substituents on the carbon atoms of the double bond are on the same side of the double bond (from the German word “zusamen” - together). The designations E- and Z- are placed before the name of the compound according to the IUPAC nomenclature and are enclosed in brackets (designation cis- And trance- is not enclosed in parentheses). For example:

Arena nomenclature
The simplest aromatic hydrocarbon with the composition C6HbC6Hb has a trivial name benzene. All other hydrocarbons in this series can be called substituted benzene derivatives, or have their own trivial names. Moreover, according to the tradition established in the Russian language, almost all trivial names of benzene homologues also have the ending -ol. For example: C6H5CH3C6H5CH3 - methylbenzene, or toluene; C6H4(CH3)2C6H4(CH3)2 - dimethylbenzene, or xylene; C6H5CH(CH3)2C6H5CH(CH3)2 - isopropylbenzene, or cymene. As an exception, C6H3(CH3)3C6H3(CH3)3 or 1, 3, 5-trimethylbenzene is called mesitylene. According to IUPAC rules, all names of aromatic hydrocarbons are characterized by the ending -en. Accordingly: benzene, toluene, xylene, cymene, styrene, etc.
In practice, to form the names of two or more substituted single-core arenas The following options are most often used:
1. The name is based on the trivial name arene (toluene, styrene, etc.); Russian letters (o-, m-, p-) or Latin (o-, m-, p-) are used to indicate the location of the side chains. , which means the ortho, meta, or para positions of the benzene ring. Alkyl radicals or functional groups are named according to the IUPAC nomenclature: methyl-, ethyl-, isopropyl-, amino-, hydroxo-, nitro-, etc. Often such rules are used to form the names of aromatic compounds of other classes - aminobenzenes, phenols, etc., containing various substituents.

2. Less commonly used are names based on the word “benzene”, and the location of radical substituents is indicated by numbers. When naming more complex benzene derivatives, as in the case of alicyclic compounds, from the possible orders, choose the one at which the sum of the digits of the substituent numbers is the smallest. However, there are no generally accepted rules for the numbering order of the atoms of the benzene ring. According to the Geneva nomenclature, number 1 is assigned to the atom of the substituent to which the substituent atom with the lowest atomic weight is directly bonded (for example, if there is -Cl and -OH in the nucleus, number 1 is assigned to the atom bearing -OH, but in the presence of -NO2NO2 and -OH - atom bearing -NO2; in substituted derivatives of benzene homologs, the beginning of numbering is determined by the simplest side chain. multi-core arenas The rules of IUPAC nomenclature establish a list of names that form the basis for the nomenclature of condensed multinuclear carbocyclic systems, the rules for the orientation of their formulas and the order of numbering of atoms. The nomenclature uses trivial names (naphthalene, phenanthrene, anthracene) indicating the location of the substituents. For example, for naphthalene derivatives, both rules described above for mononuclear arenes can also be used:
The nature of the bonds in the benzene molecule The benzene molecule contains a system of conjugated bonds. All six carbon atoms of the cyclic benzene molecule C6H6 lie in the same plane. There are σ bonds between carbon atoms in the plane of the ring; Each carbon atom has the same bonds with hydrogen atoms. Carbon atoms spend three electrons to make these bonds. Clouds of fourth valence electrons of carbon atoms, shaped like figures of eight, are located perpendicular to the plane of the benzene molecule. Each such cloud overlaps equally with the electron clouds of neighboring carbon atoms. In the benzene molecule, not three separate π-bonds are formed, but a single π-electron system of six electrons, common to all carbon atoms. The bonds between the carbon atoms in the benzene molecule are exactly the same. All bonds between carbon atoms in benzene are equivalent, which determines the characteristic properties of the benzene ring. This is most accurately reflected by the structural formula of benzene in the form of a regular hexagon with a circle inside. (The circle symbolizes the equivalence of bonds between carbon atoms.) However, Kekulé’s formula indicating double bonds is also often used.
Acid-base properties of alcohols. Metal alcoholates, their basic and nucleophilic properties. Nucleophilic substitution reactions involving alcohols. Examples of biologically important nucleophilic substitution reactions involving phosphoric acid esters.
alcohols are weak Brønsted OH acids and hard Pearson acids. Alcohols are close to water in acidity. The acidic properties of alcohols are determined by the ability to protonate the hydrogen atom of the hydroxyl group. The latter is determined not only by the difference in electronegativity between the oxygen (3.5) and hydrogen (2.1) atoms, but also by the nature of the radical. Methanol (pK a = 15.5) is a slightly stronger acid than water (pK a = 15.7), but most alcohols are weaker acids than water. The reason for this is steric hindrances that interfere with the solvation of the resulting alkoxide anion in branched alcohols. Solvation stabilizes the alkoxide anion and therefore enhances the acidic properties. Reactions involving a nucleophilic center. The high electronegativity of the oxygen atom (3.5 on the Pauling scale), which is the main center, allows us to consider alcohols as weak n-Brønsted bases and hard Pearson bases. Alcohols are capable of forming oxonium salts only with strong protic acids and hard Pearson acids (boron fluoride , zinc chloride, etc. Thus, alcohols have weak acidic and weak basic properties, i.e. they are amphiprotic compounds. At a sufficiently high temperature and in the absence of a good nucleophile, protonated alcohols are capable of reaction, i.e. to the dehydration reaction. Being rigid bases, due to the low polarizability and high electronegativity of the oxygen atom, alcohols are weak nucleophiles. Bronsted acids protonate the oxygen atom of the hydroxy group.
42. Intra- and intermolecular dehydration of alcohols .: Dehydration of alcohols can be carried out in two directions: intramolecular and intermolecular. The direction of dehydration depends on the nature of the alcohol and the reaction conditions. During intramolecular dehydration of alcohol, an unsaturated ethylene hydrocarbon is formed, and as a result of intermolecular dehydration, an ether is formed. Thus, when alcohols are heated with such water-removing substances as concentrated H 2 SO 4, H 3 PO 4, anhydrous oxalic acid, aluminum oxide, etc., unsaturated compounds of the ethylene series are formed. The reactivity of alcohols to dehydration, that is, to the formation of which ethylene compounds, changes in the following order: tertiary alcohols > secondary alcohols > primary alcohols. Some tertiary alcohols dehydrate so easily that they can only be distilled if they are prevented from entering even laboratory air, which contains trace amounts of acid vapor .Dehydration of alcohols in the presence of concentrated H 2 SO 4, depending on the temperature and the ratio of the volumes of alcohol and acid, can be carried out with the formation of different products. For example, ethyl alcohol at 105 o C forms an acid ester with sulfuric acid - ethyl sulfuric acid (reaction 1). With an excess of alcohol and a higher temperature (130–140 o C), intermolecular dehydration occurs, the main product of which is diethyl ether (ether; reaction 2). At temperatures above 160 o C, ethyl sulfuric acid decomposes to form ethylene (reaction 3):
43. Oxidation of primary and secondary alcohols. Alcohols at 300-400 o C and in the presence of copper and other catalysts are oxidized by atmospheric oxygen. Oxidizing agents such as KMnO 4 and chromium mixture oxidize alcohols already at room temperature. Depending on what kind of alcohol it is - primary, secondary or tertiary - different products are formed during oxidation.
Primary alcohols, when oxidized, give aldehydes with the same number of carbon atoms as in the molecule of the original alcohol. Under these conditions, aldehydes can be oxidized to carboxylic acids. To avoid further oxidation, aldehydes must be quickly removed from the reaction mixture
Primary alcohols can also be oxidized into aldehydes with finely crushed copper. Heated to 280-300 o C. Under these conditions, two hydrogen atoms are split off from the alcohol molecule and a carbon-oxygen double bond (>C=O) appears in the organic molecule that is formed. This transformation of alcohols is called dehydrogenation:
![]()
Secondary alcohols during oxidation, as well as during dehydrogenation, turn into ketones:
Tertiary alcohols are quite difficult to oxidize with simultaneous breaking of the carbon chain of their molecules and the formation of a mixture of carboxylic acids and ketones. This oxidation of these alcohols is due to the fact that under the conditions of the oxidation reaction they are dehydrated and converted into ethylene hydrocarbons, which, in the presence of a strong oxidizing agent, are oxidized with the rupture of the molecule at the site of the double C=C bond
Sulfonation
Sulfonation of phenol is carried out by heating with concentrated sulfuric acid. The temperature of the reaction decisively determines the structure of the resulting hydroxybenzenesulfonic acids. ortho-Isomer, the rate of formation of which is higher than pair-isomer is the dominant product if the reaction temperature does not exceed 100 °C. It is called a kinetic product. In contrast, at higher temperatures the main product is pair-isomer, the rate of formation of which is lower, but it has high thermodynamic stability. The reaction of electrophilic aromatic sulfonation is reversible when heated ortho-hydroxybenzenesulfonic acids, with sulfuric acid above 100 °C are obtained pair-isomer is a product of thermodynamic control of the reaction. Alkylation.Unlike the alkylation of phenol at the hydroxy group, which occurs in an alkaline medium, the introduction of alkyl substituents into the aromatic ring of phenol occurs under the action of haloalkanes, alcohols or alkenes in the presence of catalysts - mineral acids or Lewis acids (Friedel-Crafts reaction). Picric acid. The presence of three nitro groups in the nucleus sharply increases the acidity of the phenolic group. Picric acid, unlike phenol, is already a fairly strong acid. The presence of three nitro groups makes picric acid explosive and is used to prepare melinite. To obtain mononitrophenols, it is necessary to use dilute nitric acid and carry out the reaction at low temperatures: A mixture of o- and p-nitrophenols with a predominance of the o-isomer is obtained. This mixture is easily separated due to the fact that only the o-isomer is volatile with water vapor. The greater volatility of o-nitrophenol is explained by the formation of an intramolecular hydrogen bond, while in the case of p-nitrophenol an intermolecular hydrogen bond occurs.
47. Carboxylation of alkali metal phenolates. Salicylic acid. Phenolocarboxylic acids are obtained by reacting alkali metal phenolates with carbon (IV) oxide. Salicylic (o-hydroxybenzoic) acid is one of the most important phenolocarboxylic acids. It is used for the production of drugs (sodium salicylate, acetylsalicylic acid, phenyl salicylate, methyl salicylate), in the synthesis of dyes, in the production of aromatic substances (esters), for the production of coumarin, etc. Stages of salicylic acid production: 1) production of anhydrous sodium phenolate:
![]()
2) carboxylation of sodium carbon (IV) phenolate oxide:
![]()
Phenol as a by-product is distilled off; 3) decomposition of crude sodium salicylate:
![]()
Sparingly soluble salicylic acid precipitates;
4) separation and purification of salicylic acid. The resulting technical salicylic acid contains up to 99% pure product. Salicylic acid intended for the production of medicinal substances must be purified by sublimation.
48. Oxidation of phenols. The increased electron density in the phenol core makes it sensitive to the effects of oxidizing agents. Depending on the nature of the oxidizing agent and the reaction conditions, various oxidation products of phenol are formed.1) When phenol is oxidized with hydrogen peroxide at
In the absence of an iron catalyst, ortho-benzoquinone is obtained through the intermediate formation of pyrocatechol:
![]()
2) Strong oxidizing agents, such as chromium mixture (K 2 Cr 2 O 7 + H 2 SO 4), bromates (KBrO 3, H 2 SO 4) oxidize phenol to para-benzoquinone through the intermediate formation of hydroquinone:
![]()
3) With a more energetic action of oxidizing agents, the benzene ring is destroyed. Due to the tendency to oxidize, phenols can become colored when stored in air.
49. Quinones and their biological role. Quinones are six-membered cyclic diketones with two double bonds. The most practical of them is paraquinone, obtained by the oxidation of hydroquinone or aniline. Paraquinone is the starting product in the synthesis of hydroquinone. The arrangement of double bonds characteristic of quinone determines the color of a number of compounds. Naphthoquinones are naphthalene derivatives containing a quinoid nucleus. The most important is 1,4-naphthoquinone, which can be obtained by the oxidation of naphthalene. In a number of its properties, 1,4-naphthoquinone is similar to p-benzoquinone. It crystallizes in the form of yellow needles, is volatile, and has a pungent irritating odor. The 1,4-naphthoquinone core is the basis of vitamin K, or an antihemorrhagic vitamin (prevents the appearance of hemorrhages). Vitamin K is 2-methyl-3-phytyl-1,4-naphthoquinone. Vitamin K is found in green herbs, leaves, and vegetables. Rep. is a yellow oil, insoluble. in water; distilled in high vacuum. Some quinone derivatives play an important role in intermediate processes of biological oxidation. Anthraquinones are anthracene derivatives containing a quinoid nucleus. Anthraquinone can be easily obtained by oxidation of anthracene with nitric acid or a chromic mixture. In this case, two keto groups are formed in the molecule and the middle ring acquires the structure of a quinone. Anthraquinone is a yellow crystalline substance, which, unlike conventional quinones, is quite resistant to a number of chemical influences, in particular to oxidation. Anthrahydroquinone is an intermediate in the reduction of anthraquinone to anthracene. Anthrahydroquinone in its free form appears as brown crystals. Having two phenolic hydroxyls, anthrahydroquinone is soluble in alkalis; the resulting phenolate-type substance has a bright red color. Anthraquinone can be brominated, nitrated and sulfonated. Alizarin is a 1,2-dioxyanthraquinone. Emodins. In medical practice, preparations (tinctures, decoctions, etc.) from aloe, rhubarb, buckthorn, senna leaves, etc. are often used as laxatives. The active substances of these plants, as it turns out, are anthraquinone derivatives, namely, substituted di- and trioxy-anthraquinones, contained in plants partly in free form, partly in the form of esters and glycosides. These di- and trioxyanthraquinone derivatives are often grouped together as emodins. An example of emodins is franguloemodin, which is 3-methyl-1,6,8-trioxyanthraquinone. Franguloemo-din is found in buckthorn
50. Concept of phenolic antioxidants. Phenolic compounds in nature. Vitamin E. Flavonoids. Antioxidants (AO) or antioxidants are usually called compounds of various chemical natures that can inhibit or eliminate the free radical oxidation of organic substances by molecular oxygen. For many years, antioxidants have been widely used to extend the service life and improve the performance of polymers and fuels and lubricants, and to prevent oxidative spoilage of foods, fat-soluble vitamins, feed and cosmetics. The use of AO in these areas provides a huge economic effect and allows saving significant raw materials. Among synthetic AOs, alkylated phenols are widely used, which is due to both the comparative simplicity of their production and a set of valuable properties: high efficiency, low toxicity, versatility of action and the ability to change their properties over a wide range by varying substituents. Phenolic antioxidants (PAOs) are understood as any compounds of the Ar(OH)n type, in which one or more hydroxyl groups are connected to an aromatic ring, and the AO molecule may contain several Ar(OH)n fragments. Phenolic compounds can influence many physiological processes occurring in the human body. For example, in the composition of herbal preparations, these substances (such as coumarin, the properties of which are still poorly understood, rutin, flavonoids) stimulate the activity of the adrenal cortex, due to which the adrenal glands begin to more actively secrete hormones of the glucocorticoid group (a type of hormone secreted by the adrenal glands). They have a wide variety of biological properties. For example, phenolic compounds in the leaves of bearberry, pear, and lingonberry act as antiseptics.
Phenolocarboxylic acids are derivatives of aromatic hydrocarbons, in the molecule of which the H atoms of the benzene ring are replaced by carbo(-COOH) or hydroxyl groups (-OH). Phenolic compounds have long found their use in medicine; they are used in the treatment of neuroses and coronary insufficiency. Phenolic compounds have diuretic, sedative, choleretic and hemostatic effects. Flavanoids and bioflavonoids belong to phenols; these are yellow-red plant pigments. There are many of them in both food and medicinal plants. Bioflavonoids strengthen capillaries, act as oncoprotectors, and participate in the removal of heavy metal salts and radionuclides from the body. The group of bioflavonoids includes special substances that also exhibit P-vitamin activity and other properties of bioflavonoids, called anthocyanins. According to their chemical structure, anthocyanins are flavone glycosides. Tannins are polymeric phenolic compounds. In medicine, they are used as astringents, anti-inflammatory gastrointestinal agents. The most famous tannin is tannin. It should not be taken orally: it will cause digestive upset. Catechins (they are also classified as bioflavonoids) are derivatives of flavonols and anthocyanins. Coumarins are aromatic substances with the smell of fresh hay. Coumarins are anticoagulants, that is, they prevent rapid blood clotting, such as the coumarin derivative dicumarol. It is antivitamin K and is used for the prevention and treatment of thrombosis and thrombophlebitis.
51. Ethers. Nomenclature, classification. Acid digestion. Ethers- organic substances in which the molecules contain hydrocarbon radicals connected by an oxygen atom. This can be written as follows: R–O–R', where R and R" are the same or different radicals. Ethers are considered as derivatives of alcohols. These compounds have compound names. In this case, the name of the radicals is used (increasing molecular weight) and , in fact, the word “ether” (dimethyl ether CH3OCH3, methyl ethyl ether C2H5OCH3 and so on)
Nomenclature of ethers According to trivial nomenclature, ethers are named by the radicals associated with the oxygen atom, adding the word “ether”.
According to IUPAC nomenclature, ethers are considered alkoxyalkanes. The longest alkyl group determines the root of a word.
![]()
Ethers are low-reactive substances and are stable with respect to many reagents, but they are sensitive to oxygen and easily form explosive hydroperoxides, which cause explosions if handled carelessly.
1 . Acid cleavage of ethers
Ethers are split when heated to 120-150 o C conc. aqueous 48% HBr or HI. Phenol ethers are broken down under equally harsh conditions.
![]()
However, ethers containing a tertiary alkyl group are cleaved very easily.
The acid cleavage of ethers should be considered as a nucleophilic substitution reaction at a saturated carbon atom. Depending on the nature of the alkyl groups associated with oxygen, either the S N 1 or S N 2 mechanisms are implemented. If the ether contains primary or secondary alkyl groups, the S N 2 mechanism is implemented in which the bromide or iodide ion attacks the protonated form of the ester at the less substituted carbon atom. In this case, the cleavage is highly regioselective and, as a rule, only one of the two possible alcohols (secondary) and primary alkyl halide is formed.
Chloride and fluoride ions in water are highly solvated due to hydrogen bonds and have insufficient nucleophilicity for acid cleavage of ethers by the S N 2 mechanism.
Ethers with tertiary alkyl, benzyl or allyl groups react via the S N 1 mechanism to form a carbocation as an intermediate. These reactions occur under mild conditions, and trifluoroacetic acid can be used as an acidic agent.
From a preparative point of view, much more convenient reagents for splitting esters are BCl 3 or BBr 3 . In these cases, cleavage occurs already at -20 o C. This is especially necessary in the presence of other functional groups or when isomerization of the carbon skeleton is possible.
Formation of hydroperoxides, their detection and decomposition.
Hydroperoxides are the first molecular products of hydrocarbon oxidation. The chain link during their formation has the form:
The interaction of a peroxide radical with a hydrocarbon determines the structure of the resulting hydroperoxide and subsequent oxidation products. In this case, the usual order of changes in the reactivity of hydrogen atoms for radical reactions is observed, determined by the relative stability of the intermediate radical. As a result, the preferred site of attack of the molecule during the oxidation of alkanes becomes the -position of the side chain relative to the aromatic ring, and for olefins, the alkyl position. In addition, for hydrocarbons of all classes, the known sequence in changing the ability to replace hydrogen atoms located at different carbon atoms is valid (tertiary secondary primary).
Hydroperoxides are fairly unstable compounds that transform into other products upon oxidation. Hydroperoxides, when decomposed under the influence of elevated temperature or oxidation catalysts, produce alcohols and carbonyl compounds. This decomposition may have a molecular mechanism, but in the developed oxidation process the products are formed mainly in a chain way. When producing alcohols, the chain link is as follows:
Ketones are formed from secondary hydroperoxides through the radical hydroperoxide stage:
Tertiary hydroperoxides during a chain transformation produce, in addition to alcohol with the same number of carbon atoms, also alcohol and ketone with a smaller number of carbon atoms due to the destruction of the carbon-carbon bond:
![]()
Features of the properties of arylamines. Electrophilic substitution reactions in the benzene ring of arylamines and their derivatives. Diazotization reactions, aryldiazonium salts. Reactions of aryldiazonium salts with and without nitrogen evolution.
Arylamines are characterized by reactions involving the nitrogen atom and reactions involving the carbon atoms of the aromatic ring. Basicity. Aromatic amines are basic in nature. However, they are weaker than fatty amines and even weaker than ammonia. The decrease in basicity is due to the conjugation of the lone pair of electrons of the nitrogen atom with the l:-electron system of the aromatic nucleus. Aniline does not form a salt with H2COg. The basicity of arylamines is significantly influenced by substituents on the benzene ring. Electron-donating substituents increase basicity, and electron-withdrawing substituents decrease it. During the transition from primary to tertiary, the basicity of aromatic amines decreases.
Electrophilic substitution in the benzene ring.
In electrophilic substitution reactions in the benzene ring, a hydrogen atom is replaced by an electrophilic reagent while maintaining the aromatic character of the original compound.
Arenediazonium salts are formed by the reaction of primary aromatic amines with nitrous acid. In industry, arendiazonium salts are widely used to obtain a variety of azo dyes of all colors and shades. For this reason, diazotization is one of the most important and best-studied reactions in organic chemistry.
Diazotization of primary aromatic amines is described by the following summary equation:
ArNH 2 + NaNO 2 + 2 HClArN + =N Cl - + NaCl + 2 H 2 O
Reactions with nitrogen release. When acidic solutions of diazonium salts are boiled, nitrogen is released and phenols are obtained. Conversion of diazonium salts without releasing nitrogen. The reactions of this group make possible the transition from diazo compounds to azo compounds (azobenzene derivatives). Organic substances of this class underlie one of the sections of the industry that produces synthetic dyes from products extracted from coal tar. All azo dyes are obtained using the so-called diazonium salt coupling reaction.
59.Carbonyl compounds. Classification, nomenclature and isomerism of carbonyl compounds. Organic compounds whose molecule contains a carbonyl group >C=O are called carbonyl compounds, or oxo compounds. Carbonyl compounds are divided into two large groups - aldehydes and ketones.
Theories of acids and bases
Theories of acids and bases- a set of fundamental physical and chemical concepts that describe the nature and properties of acids and bases. They all introduce definitions of acids and bases - two classes of substances that react with each other. The task of the theory is to predict the products of the reaction between an acid and a base and the possibility of its occurrence, for which quantitative characteristics of the strength of the acid and base are used. The differences between the theories lie in the definitions of acids and bases, the characteristics of their strength and, as a consequence, in the rules for predicting the reaction products between them. They all have their own area of applicability, which areas partially overlap.
Acid-base interactions are extremely common in nature and are widely used in scientific and industrial practice. Theoretical ideas about acids and bases are important in the formation of all conceptual systems of chemistry and have a diverse influence on the development of many theoretical concepts in all major chemical disciplines.
Based on the modern theory of acids and bases, such branches of chemical sciences as the chemistry of aqueous and non-aqueous electrolyte solutions, pH-metry in non-aqueous media, homo- and heterogeneous acid-base catalysis, the theory of acidity functions and many others have been developed.
Evolution of ideas about acid-base interactions
Scientific ideas about the nature of acids and bases began to take shape at the end of the 18th century. In the works of A. Lavoisier, acidic properties were associated with the presence of oxygen atoms in the composition of the substance. The then known mineral and organic acids actually contained oxygen. This hypothesis quickly showed its inconsistency when, thanks to the work of G. Davy and J. Gay-Lussac, a number of oxygen-free acids (for example, hydrogen halides, hydrocyanic acids) became known, while many oxygen-containing compounds do not exhibit acidic properties.
From the beginning of the 19th century, acids began to be considered substances capable of interacting with metals with the release of hydrogen (Yu. Liebig, 1839). Around the same time, J. Berzelius put forward an idea that explained the acid-base properties of substances by their electrical “dualistic” nature. Thus, he classified electronegative oxides of nonmetals and some metals (for example, chromium, manganese, etc.) as acids, and considered electropositive metal oxides as bases. Thus, Berzelius regards acidity or basicity as a functional rather than an absolute property of a compound. Berzelius pioneered the attempt to quantify and predict the strength of acids and bases.
With the advent of the theory of electrolytic dissociation by S. Arrhenius (1887), the possibility arose of describing acid-base properties based on electrolyte ionization products. Thanks to the work of W. Ostwald, the theory was developed for weak electrolytes.
At the beginning of the 20th century. American chemists G. Cady, E. Franklin and C. Kraus created the theory of solvo systems, which extended the provisions of the Arrhenius-Oswald theory to all solvents capable of self-dissociation.
Modern theories of acids and bases are based on the ideas of J. Brønsted and G. Lewis. There are quite successful attempts to create generalized theories (M. Usanovich, 1939), but they are not widely used.
Liebig's hydrogen theory
Definitions. An acid is a substance that can react with a metal to release hydrogen. The concept of "foundation" is absent in this theory.
Reaction products. When an acid reacts with a metal, a salt and hydrogen are formed.
Examples. Acid -- HCl.
Reaction 2HCl + Zn = ZnCl 2 + H 2
Criteria for the reaction. Metals that are to the left of hydrogen in the activity series react with strong acids. The weaker the acid, the more active the metal is needed for the reaction between them. Quantitative characteristics. Since the theory is rarely used, quantitative characteristics of the acid strength (and therefore predictions of the direction of the reaction) have not been developed within the framework of this theory.
Scope of applicability. Prediction of the interaction of hydrogen-containing substances with metals in any solvents.
Specific features. According to this theory, ethyl alcohol and ammonia are weak acids, as they are capable of reacting with alkali metals:
2C 2 H 5 OH + 2Na = 2C 2 H 5 ONa + H 2
2NH 3 + 2Na = 2NaNH 2 + H 2
Arrhenius-Ostwald theory of electrolytic dissociation
Main article: Electrolytic dissociation theory
For acid HA K = ·/
For MOH base K = ·/
For a reaction to take place between an acid and a base, the product of their dissociation constants must be greater than 10 -14 (the ionic product of water).
Scope of applicability. It quite satisfactorily describes the reactions of fairly strong acids and bases with each other and the properties of their aqueous solutions. Based on ideas about the degree and dissociation constant, the division of electrolytes into strong and weak was established, and the concept of hydrogen index was introduced, the extension of which to alkaline media requires, however, additional assumptions (the introduction of the ionic product of water).
The theory can be used to describe the hydrolysis of salts and the reaction of acids and bases with salts, but this requires a very cumbersome apparatus - the proton theory (see below) is much more convenient.
The applicability of the Arrhenius-Ostwald theory is limited to aqueous solutions. in addition, it does not explain the presence of the basic properties of ammonia, phosphine and other compounds that do not contain hydroxo groups.
Brønsted-Lowry proton theory
Main article: Protolytic theory of acids and bases
Comparison of models
acid-base interaction
according to Lewis and Bronsted
Protolytic (proton) theory of acids and bases was proposed in 1923 independently by the Danish scientist J. Brønsted and the English scientist T. Lauri. In it, the concept of acids and bases was combined into a single whole, manifested in the acid-base interaction: A B + H + (A - acid, B - base). According to this theory, acids are molecules or ions that can be proton donors in a given reaction, and bases are molecules or ions that add protons (acceptors). Acids and bases are collectively called protolytes.
The essence of acid-base interaction is the transfer of a proton from an acid to a base. In this case, the acid, having transferred a proton to the base, itself becomes a base, since it can again attach a proton, and the base, forming a protonated particle, becomes an acid. Thus, in any acid-base interaction, two pairs of acids and bases are involved, called by Brønsted conjugated: A1 + B2 A2 + B1.
The same substance, depending on the conditions of interaction, can be both an acid and a base (amphoteric). For example, water, when interacting with strong acids, is a base: H 2 O + H + H 3 O +, and when reacting with ammonia, it becomes an acid: NH 3 + H 2 O NH 4 + + OH − .
Solvosystem theory
Main article: Solvosystem theory
The theory of solvo systems is an extension of the Arrhenius-Ostwald theory to other ionic (in particular, protic) solvents. Proposed by American chemists G. Cady, E. Franklin and C. Kraus
Definitions. An ionic solvent is a solvent that self-dissociates into a cation and an anion. The cation is called a lyonium ion, and the anion is called a lyate ion. A protic solvent is a solvent capable of autoprotolysis, that is, the transfer of the H + ion from one molecule to another:
2HL ↔ H 2 L + + L -
These are solvents containing a fairly polar bond involving hydrogen and a lone electron pair on some other non-metal (most often nitrogen, oxygen or fluorine).
Note: in this definition the proton theory is “hardwired”, because autoprotolysis is an acid-base reaction according to Breasted-Lowry. It also contains the Lewis theory, since it explains the reasons for the formation of lyonium ions.
The H 2 L + ion is called the lyonium ion, and the L - ion is called the lyate ion.
Acids are substances that form a lyonium ion in a given solvent.
Bases are substances that form a lyate ion in a given solvent.
Salts are substances that dissociate in a given solvent to form a cation and an anion that are not lyonium or lyate.
Reaction products. When an acid reacts with a base (neutralization reaction), a salt and a solvent are formed.
Examples.
Quantitative characteristics and criteria for the reaction The strengths of acids and bases are characterized by their dissociation constant.
Dissociation constants depend on the solvent. Protic solvents with high autodissociation constants (“acidic solvents”, for example HF) differentiate acids (in them the acids become weak and differ in strength) but neutralize bases (all bases become strong, turning into the lyate ion). Protic solvents with low autodissociation constants (“basic solvents, such as NH 3 ) differentiate bases but neutralize acids (which become strong, becoming lyonium).
The reaction goes from strong acids to weak ones.
Scope of applicability. Allows you to predict acid-base reactions in any solvent. Control of acid-base processes using a solvent. Extends the concept of pH as the concentration of lyonium ions to non-aqueous solutions. Describes the basic properties of substances that do not contain OH groups.
However, for many problems the theory is too cumbersome.
Specific features Some acid-base reactions in this theory can be turned upside down, for example
KOH (acid) + HCl (base) = KCl (solvent) + H 2 O (salt)
Lewis electron theory
Main article: Lewis theory
In the theory of Lewis (1923), based on electronic concepts, the concept of acid and base was further expanded. A Lewis acid is a molecule or ion that has vacant electron orbitals, as a result of which it is capable of accepting electron pairs. These are, for example, hydrogen ions - protons, metal ions (Ag +, Fe 3+), oxides of some non-metals (for example, SO 3, SiO 2), a number of salts (AlCl 3), as well as substances such as BF 3, Al 2 O3. Lewis acids that do not contain hydrogen ions are called aprotic acids. Protic acids are considered as a special case of the class of acids. A Lewis base is a molecule or ion capable of donating electron pairs: all anions, ammonia and amines, water, alcohols, halogens. Examples of chemical reactions between Lewis acids and bases:
- AlCl 3 + Cl − → AlCl 4 −
- BF 3 + F − → BF 4 −
- PCl 5 + Cl − → PCl 6 − .
Usanovich's general theory
The most general theory of acids and bases was formulated by M. Usanovich in 1939. The theory is based on the idea that any acid-base interaction is a salt formation reaction. According to this theory " an acid is a particle that can remove cations, including a proton, or add anions, including an electron. Base - a particle that can accept a proton and other cations or donate an electron and other anions"(wording 1964). Unlike Lewis, Usanovich bases the concepts of “acid” and “base” on the sign of the particle’s charge, and not on the structure of the electron shell.
Usanovich's theory actually abolishes one of the fundamental principles of classical chemistry - the concept of classes of acids and bases: “ acids and bases are not classes of compounds; acidity and basicity are functions of a substance. Whether a substance is an acid or a base depends on the partner» .
The disadvantages of Usanovich’s theory include its too general nature and insufficiently clear definition of the formulation of the concepts “acid” and “base”. Disadvantages also include the fact that it does not describe nonionic acid-base transformations. Finally, it does not allow quantitative predictions