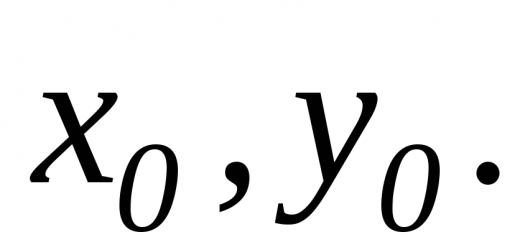Während Atomspektren aus einzelnen Linien bestehen, scheinen Molekülspektren, wenn sie mit einem Instrument mit mittlerem Auflösungsvermögen beobachtet werden, aus einzelnen Linien zu bestehen (siehe Abb. 40.1, die einen Ausschnitt aus dem Spektrum einer Glimmentladung in Luft zeigt).


Beim Einsatz hochauflösender Instrumente stellt man fest, dass die Banden aus einer Vielzahl eng beieinander liegender Linien bestehen (siehe Abb. 40.2, die die Feinstruktur einer der Banden im Spektrum der Stickstoffmoleküle zeigt).
Die Spektren von Molekülen werden ihrer Natur nach als Streifenspektren bezeichnet. Abhängig von der Änderung der Energiearten (elektronisch, schwingend oder rotatorisch), die die Emission eines Photons durch ein Molekül bewirken, werden drei Arten von Bändern unterschieden: 1) Rotationsbänder, 2) Schwingungs-Rotationsbänder und 3) Elektronisch-Schwingungsbänder. Streifen in Abb. 40.1 gehören zum elektronischen Schwingungstyp. Diese Art von Streifen zeichnet sich durch das Vorhandensein einer scharfen Kante aus, die als Streifenkante bezeichnet wird. Der andere Rand eines solchen Streifens erweist sich als unscharf. Die Kanten entstehen durch die Verdichtung von Linien, die einen Streifen bilden. Rotations- und Oszillations-Rotationsbänder haben keine Kante.
Wir beschränken uns auf die Betrachtung der Rotations- und Vibrations-Rotationsspektren zweiatomiger Moleküle. Die Energie solcher Moleküle besteht aus Elektronen-, Schwingungs- und Rotationsenergien (siehe Formel (39.6)). Im Grundzustand des Moleküls haben alle drei Energiearten einen minimalen Wert. Wenn einem Molekül eine ausreichende Energiemenge zugeführt wird, geht es in einen angeregten Zustand über und emittiert dann ein Photon, indem es einen durch die Auswahlregeln zulässigen Übergang in einen der niedrigeren Energiezustände vollzieht:
(Es muss berücksichtigt werden, dass sich beide für verschiedene elektronische Konfigurationen des Moleküls unterscheiden).
Im vorherigen Absatz wurde Folgendes festgestellt
![]()
Daher ändert es sich bei schwachen Anregungen nur bei stärkeren – und erst bei noch stärkeren Anregungen ändert sich die elektronische Konfiguration des Moleküls, d. h.
Rotationsstreifen. Photonen, die Übergängen eines Moleküls von einem Rotationszustand in einen anderen entsprechen, haben die niedrigste Energie (die elektronische Konfiguration und die Schwingungsenergie ändern sich nicht):
Mögliche Änderungen der Quantenzahl werden durch die Auswahlregel (39.5) begrenzt. Daher können die Frequenzen der bei Übergängen zwischen Rotationsebenen emittierten Linien folgende Werte annehmen:
wo ist die Quantenzahl der Ebene, zu der der Übergang erfolgt (sie kann die Werte 0, 1, 2, ... haben) und
In Abb. Abbildung 40.3 zeigt ein Diagramm des Auftretens eines Rotationsbandes.


Das Rotationsspektrum besteht aus einer Reihe gleichmäßig beabstandeter Linien im sehr fernen Infrarotbereich. Durch Messen des Abstands zwischen den Linien können Sie die Konstante (40.1) bestimmen und das Trägheitsmoment des Moleküls ermitteln. Wenn man dann die Massen der Kerne kennt, kann man den Gleichgewichtsabstand zwischen ihnen in einem zweiatomigen Molekül berechnen.
Der Abstand zwischen den Lie-Linien liegt in der Größenordnung, sodass sich für die Trägheitsmomente von Molekülen Werte in der Größenordnung ergeben. Beispielsweise für ein Molekül, was entspricht.
Vibrations-Rotationsbänder. Wenn sich während des Übergangs sowohl der Schwingungs- als auch der Rotationszustand des Moleküls ändern (Abb. 40.4), ist die Energie des emittierten Photons gleich
Für die Quantenzahl v gilt die Auswahlregel (39.3), für J gilt die Regel (39.5).
Da die Photonenemission nicht nur bei und bei beobachtet werden kann. Wenn die Photonenfrequenzen durch die Formel bestimmt werden
wobei J die Rotationsquantenzahl der unteren Ebene ist, die folgende Werte annehmen kann: 0, 1, 2, ; B - Wert (40,1).
Wenn die Formel für die Photonenfrequenz die Form hat
Wo ist die Rotationsquantenzahl der unteren Ebene, die die Werte 1, 2, ... annehmen kann (in diesem Fall kann sie nicht den Wert 0 haben, da dann J gleich -1 wäre).
Beide Fälle können durch eine Formel abgedeckt werden:
Die durch diese Formel bestimmte Menge von Linien mit Frequenzen wird als Schwingungs-Rotationsband bezeichnet. Der Schwingungsanteil der Frequenz bestimmt den Spektralbereich, in dem sich das Band befindet; Der Rotationsanteil bestimmt die Feinstruktur des Streifens, also die Aufspaltung einzelner Linien. Der Bereich, in dem sich die Schwingungs-Rotationsbänder befinden, erstreckt sich von etwa 8000 bis 50000 A.
Aus Abb. 40.4 ist klar, dass das Schwingungs-Rotationsband aus einer Reihe relativ symmetrischer Linien besteht, die nur in der Mitte des Bandes einen doppelt so großen Abstand voneinander haben, da eine Linie mit Frequenz nicht erscheint.
Der Abstand zwischen den Komponenten des Schwingungs-Rotationsbandes steht mit dem Trägheitsmoment des Moleküls in derselben Beziehung wie im Fall des Rotationsbandes in Zusammenhang, so dass durch Messung dieses Abstands das Trägheitsmoment des Moleküls ermittelt werden kann gefunden.
Beachten Sie, dass in voller Übereinstimmung mit den Schlussfolgerungen der Theorie Rotations- und Vibrations-Rotationsspektren experimentell nur für asymmetrische zweiatomige Moleküle (d. h. Moleküle, die aus zwei verschiedenen Atomen bestehen) beobachtet werden. Bei symmetrischen Molekülen ist das Dipolmoment Null, was zum Verbot von Rotations- und Schwingungs-Rotations-Übergängen führt. Elektronische Schwingungsspektren werden sowohl für asymmetrische als auch für symmetrische Moleküle beobachtet.
Neben den Spektren, die der Strahlung einzelner Atome entsprechen, werden auch Spektren ganzer Moleküle beobachtet (§ 61). Molekülspektren sind viel vielfältiger und komplexer aufgebaut als Atomspektren. Hier werden verdichtete Linienfolgen beobachtet, ähnlich der Spektralreihe von Atomen, jedoch mit einem anderen Frequenzgesetz und mit Linien, die so eng beieinander liegen, dass sie zu kontinuierlichen Bändern verschmelzen (Abb. 279). Aufgrund der besonderen Natur dieser Spektren werden sie gestreift genannt.
Reis. 279. Gestreiftes Spektrum
Daneben werden Abfolgen gleichabständiger Spektrallinien und schließlich Mehrlinienspektren beobachtet, in denen sich auf den ersten Blick nur schwer Muster erkennen lassen (Abb. 280). Es ist zu beachten, dass wir bei der Untersuchung des Spektrums von Wasserstoff immer eine Überlagerung des molekularen Spektrums von Ha mit dem Atomspektrum haben und besondere Maßnahmen ergriffen werden müssen, um die Intensität der von einzelnen Wasserstoffatomen emittierten Linien zu erhöhen.

Reis. 280. Molekulares Spektrum von Wasserstoff
Aus quantentechnischer Sicht wird, wie im Fall von Atomspektren, jede Linie des Molekülspektrums emittiert, wenn ein Molekül von einem stationären Energieniveau in ein anderes übergeht. Aber im Fall eines Moleküls gibt es noch viele weitere Faktoren, von denen die Energie des stationären Zustands abhängt.
Im einfachsten Fall eines zweiatomigen Moleküls setzt sich die Energie aus drei Teilen zusammen: 1) der Energie der Elektronenhülle des Moleküls; 2) die Energie der Schwingungen der Atomkerne, aus denen das Molekül entlang der sie verbindenden Geraden besteht; 3) die Rotationsenergie der Kerne um einen gemeinsamen Massenschwerpunkt. Alle drei Energiearten sind quantisiert, das heißt, sie können nur eine diskrete Wertereihe annehmen. Die Elektronenhülle eines Moleküls entsteht durch die Verschmelzung der Elektronenhüllen der Atome, aus denen das Molekül besteht. Als Grenzfall können energetische elektronische Zustände von Molekülen betrachtet werden
ein sehr starker Stark-Effekt, der durch die interatomare Wechselwirkung von Atomen entsteht, die ein Molekül bilden. Obwohl die Kräfte, die Atome zu Molekülen verbinden, rein elektrostatischer Natur sind, erwies sich ein korrektes Verständnis der chemischen Bindung nur im Rahmen der modernen wellenmechanischen Quantentheorie als möglich.
Es gibt zwei Arten von Molekülen: homöopolare und heteropolare. Mit zunehmendem Abstand zwischen den Kernen zerfallen homöopolare Moleküle in neutrale Teile. Hämopolare Moleküle umfassen Moleküle. Heteropolare Moleküle zerfallen mit zunehmendem Abstand zwischen den Kernen in positive und negative Ionen. Ein typisches Beispiel für heteropolare Moleküle sind beispielsweise Salzmoleküle usw. (Bd. I, § 121, 130, 1959; in der vorherigen Ausgabe § 115 und 124 usw. II, § 19, 22, 1959; in die vorherige Ausgabe § 21 und 24).
Die Energiezustände der Elektronenwolke eines homöopolaren Moleküls werden zu einem großen Teil durch die Welleneigenschaften der Elektronen bestimmt.
Betrachten wir ein sehr grobes Modell des einfachsten Moleküls (ein ionisiertes Wasserstoffmolekül, das zwei potenzielle „Löcher“ darstellt, die in geringem Abstand voneinander liegen und durch eine „Barriere“ getrennt sind (Abb. 281).

Reis. 281. Zwei mögliche Löcher.

Reis. 282. Wellenfunktionen eines Elektrons bei entfernten „Brunnen“.
Jedes der „Löcher“ repräsentiert eines der Atome, aus denen das Molekül besteht. Bei einem großen Abstand zwischen den Atomen hat das Elektron in jedem von ihnen quantisierte Energiewerte, die den stehenden Elektronenwellen in jedem der „Wells“ separat entsprechen (§ 63). In Abb. In Abb. 282, a und b sind zwei identische Wellenfunktionen dargestellt, die den Zustand von Elektronen beschreiben, die sich in isolierten Atomen befinden. Diese Wellenfunktionen entsprechen demselben Energieniveau.
Wenn sich Atome zu einem Molekül zusammenschließen, wird die „Barriere“ zwischen den „Löchern“ „transparent“ (§ 63), weil ihre Breite der Länge der Elektronenwelle entspricht. Infolgedessen gibt es
Austausch von Elektronen zwischen Atomen durch eine „Barriere“, und es macht keinen Sinn, über die Zugehörigkeit eines Elektrons zu dem einen oder anderen Atom zu sprechen.
Die Wellenfunktion kann nun zwei Formen haben: c und d (Abb. 283). Fall c kann näherungsweise als Ergebnis der Addition der Kurven a und b (Abb. 282) betrachtet werden, Fall als Differenz zwischen a und b, aber die Energien, die den Zuständen c und d entsprechen, sind einander nicht mehr genau gleich. Die Energie des Zustands ist etwas geringer als die Energie des Zustands. Somit entstehen aus jeder atomaren Ebene zwei molekulare elektronische Ebenen.

Reis. 283. Wellenfunktionen eines Elektrons bei engen „Töpfen“.
Bisher haben wir vom Ion eines Wasserstoffmoleküls gesprochen, das über ein Elektron verfügt. Ein neutrales Wasserstoffmolekül hat zwei Elektronen, was dazu führt, dass die relativen Positionen ihrer Spins berücksichtigt werden müssen. Gemäß dem Pauli-Prinzip scheinen sich Elektronen mit parallelen Spins gegenseitig zu „vermeiden“, daher ist die Wahrscheinlichkeitsdichte, jedes Elektron zu finden, gemäß Abb. verteilt. 284, a, d.h. Elektronen befinden sich am häufigsten außerhalb der Lücke zwischen den Kernen. Daher kann mit parallelen Spins kein stabiles Molekül gebildet werden. Im Gegenteil, antiparallele Spins entsprechen der höchsten Wahrscheinlichkeit, beide Elektronen innerhalb der Lücke zwischen den Kernen zu finden (Abb. 294, b). In diesem Fall zieht die negative elektronische Ladung beide positiven Kerne an und das gesamte System bildet als Ganzes ein stabiles Molekül.
In heteropolaren Molekülen ist das Verteilungsmuster der Elektronenladungsdichte viel klassischer. In der Nähe eines der Kerne gruppiert sich ein Elektronenüberschuss, während in der Nähe des anderen ein Elektronenmangel herrscht. So entstehen im Molekül zwei Ionen, positiv und negativ, die sich gegenseitig anziehen: zum Beispiel und
Die Symbolik der elektronischen Zustände von Molekülen weist viele Ähnlichkeiten mit der Atomsymbolik auf. Natürlich spielt in einem Molekül die Richtung der Achse, die die Kerne verbindet, die Hauptrolle. Hier wird die Quantenzahl A eingeführt, analog zu I im Atom. Die Quantenzahl charakterisiert den Absolutwert der Projektion des resultierenden Bahnimpulses der Elektronenwolke des Moleküls auf die Achse des Moleküls.
Zwischen den Werten und Symbolen molekularer elektronischer Zustände besteht eine ähnliche Entsprechung wie bei Atomen (§ 67):

Der Absolutwert der Projektion des resultierenden Spins der Elektronenwolke auf die Achse des Moleküls wird durch die Quantenzahl 2 charakterisiert, und die Projektion des gesamten Rotationsmoments der Elektronenhülle wird durch die Quantenzahl charakterisiert. Offensichtlich ist
Die Quantenzahl ähnelt der inneren Quantenzahl eines Atoms (§59 und 67).

Reis. 284. Wahrscheinlichkeitsdichte, ein Elektron an verschiedenen Punkten eines Moleküls zu finden.
Genau wie Atome weisen Moleküle eine Vielfältigkeit auf, die durch unterschiedliche Ausrichtungen des resultierenden Spins relativ zum resultierenden Bahnimpuls verursacht wird.
Unter Berücksichtigung dieser Umstände werden die elektronischen Zustände von Molekülen wie folgt geschrieben:
Dabei ist 5 der Wert des resultierenden Spins und bedeutet eines der Symbole oder A, das verschiedenen Werten der Quantenzahl A entspricht. Beispielsweise ist der Normalzustand eines Wasserstoffmoleküls 2, der Normalzustand eines Hydroxyls Molekül ist der Normalzustand eines Sauerstoffmoleküls. Bei Übergängen zwischen verschiedenen elektronischen Zuständen gelten folgende Auswahlregeln: .
Die mit Schwingungen von Kernen verbundene Schwingungsenergie eines Moleküls wird unter Berücksichtigung der Welleneigenschaften von Kernen quantisiert. Unter der Annahme, dass die Kerne in einem Molekül durch eine quasielastische Kraft gebunden sind (die potentielle Energie eines Teilchens ist proportional zum Quadrat der Verschiebung, § 63), erhalten wir aus der Schrödinger-Gleichung die folgenden zulässigen Werte der Schwingung Energie dieses Systems (harmonisch
Oszillator):
![]()
wo ist die Frequenz der natürlichen Schwingungen der Kerne, wie üblich bestimmt (Bd. I, § 57, 1959; in der vorherigen Ausgabe § 67):
![]()
wo ist die reduzierte Masse der Kerne; Massen beider Kerne; quasielastische Konstante eines Moleküls; Quantenzahl gleich Aufgrund der großen Masse liegt die Frequenz im Infrarotbereich des Spektrums.

Reis. 285. Niveaus der Schwingungsenergie eines Moleküls.
Die quasielastische Konstante hängt von der Konfiguration der Elektronenhülle ab und ist daher für verschiedene elektronische Zustände des Moleküls unterschiedlich. Diese Konstante ist umso größer, je stärker das Molekül ist, also je stärker die chemische Bindung ist.
Formel (3) entspricht einem System gleichmäßig verteilter Energieniveaus, deren Abstand zwischen ihnen beträgt. Tatsächlich beginnen bei großen Amplituden der Kernschwingungen bereits Abweichungen der Rückstellkraft vom Hookeschen Gesetz zu wirken. Dadurch rücken die Energieniveaus näher zusammen (Abb. 285). Bei ausreichend großen Amplituden zerfällt das Molekül in Teile.
Bei einem harmonischen Oszillator sind Übergänge nur bei zulässig, was der Emission oder Absorption von Licht der Frequenz entspricht. Aufgrund von Abweichungen von der Harmonik treten Übergänge auf, die entsprechen
Gemäß der Quantenbedingung für Frequenzen (§ 58) sollten in diesem Fall Obertöne auftreten, die in den Spektren von Molekülen beobachtet werden.
Schwingungsenergie ist eine relativ kleine Ergänzung zur Energie der Elektronenwolke eines Moleküls. Schwingungen von Kernen führen dazu, dass sich jede elektronische Ebene in ein System enger Ebenen verwandelt, die unterschiedlichen Werten der Schwingungsenergie entsprechen (Abb. 286). Damit ist die Komplexität des Systems der Energieniveaus eines Moleküls noch nicht erschöpft.

Reis. 286. Addition von Schwingungs- und elektronischer Energie eines Moleküls.
Es ist auch notwendig, die kleinste Komponente der molekularen Energie zu berücksichtigen – die Rotationsenergie. Die zulässigen Werte der Rotationsenergie werden nach der Wellenmechanik nach dem Prinzip der Drehmomentquantisierung bestimmt.
Nach der Wellenmechanik ist das Drehmoment (§ 59) eines jeden quantisierten Systems gleich
![]()
In diesem Fall ersetzt es und ist gleich 0, 1, 2, 3 usw.
Kinetische Energie eines rotierenden Körpers im vorherigen. Hrsg. § 42) wird sein
![]()
wobei das Trägheitsmoment co die Winkelgeschwindigkeit der Rotation ist.
Aber andererseits ist das Drehmoment gleich. Daher erhalten wir:
oder wenn wir den Ausdruck (5) ersetzen, finden wir schließlich:
![]()
In Abb. 287 zeigt die Rotationsniveaus des Moleküls; Im Gegensatz zu Schwingungs- und Atomebenen nimmt der Abstand zwischen Rotationsebenen mit zunehmender Geschwindigkeit zu. Übergänge zwischen Rotationsebenen sind zulässig und es werden Linien mit Frequenzen ausgesendet
![]()
wo Evrash entspricht entspricht
Formel (9) gibt für Frequenzen an

Reis. 287. Niveaus der Rotationsenergie eines Moleküls.
Wir erhalten äquidistante Spektrallinien, die im fernen Infrarotbereich des Spektrums liegen. Die Messung der Frequenzen dieser Linien ermöglicht die Bestimmung des Trägheitsmoments des Moleküls. Es stellte sich heraus, dass die Trägheitsmomente von Molekülen in der Größenordnung liegen. Es ist zu beachten, dass das Trägheitsmoment I selbst aufgrund der Aktion
Die Zentrifugalkräfte nehmen mit zunehmender Rotationsgeschwindigkeit des Moleküls zu. Das Vorhandensein von Rotationen führt zur Aufspaltung jedes Schwingungsenergieniveaus in eine Reihe nahe beieinander liegender Unterniveaus, die unterschiedlichen Werten der Rotationsenergie entsprechen.
Wenn ein Molekül von einem Energiezustand in einen anderen übergeht, können sich alle drei Energiearten des Moleküls gleichzeitig ändern (Abb. 288). Dadurch erhält jede Spektrallinie, die bei einem Elektronen-Schwingungs-Übergang emittiert würde, eine feine Rotationsstruktur und verwandelt sich in ein typisches Molekülband.

Reis. 288. Gleichzeitige Änderung aller drei Energiearten eines Moleküls
Solche Bänder gleichmäßig verteilter Linien werden in Dampf und Wasser beobachtet und liegen im fernen Infrarotbereich des Spektrums. Sie werden nicht im Emissionsspektrum dieser Dämpfe beobachtet, sondern in ihrem Absorptionsspektrum, da die Frequenzen, die den Eigenfrequenzen der Moleküle entsprechen, stärker absorbiert werden als andere. In Abb. 289 zeigt eine Bande im Dampfabsorptionsspektrum im nahen Infrarotbereich. Dieses Band entspricht Übergängen zwischen Energiezuständen, die sich nicht nur in der Rotationsenergie, sondern auch in der Schwingungsenergie (bei konstanter Energie der Elektronenhüllen) unterscheiden. In diesem Fall ändern sich und und Ecol gleichzeitig, was zu großen Energieänderungen führt, d. h. die Spektrallinien haben eine höhere Frequenz als im ersten betrachteten Fall.
Demnach erscheinen im Spektrum im nahen Infrarotbereich liegende Linien, ähnlich denen in Abb. 289.

Reis. 289. Absorptionsbande.
Die Mitte des Bandes ( entspricht einem Übergang bei konstantem EUR; gemäß der Auswahlregel werden solche Frequenzen vom Molekül nicht emittiert. Linien mit höheren Frequenzen – kürzeren Wellenlängen – entsprechen Übergängen, bei denen die Änderung des EUR hinzugefügt wird Die Änderung. Linien mit niedrigeren Frequenzen (rechte Seite) entsprechen der umgekehrten Beziehung: Änderung der Rotationsenergie hat das umgekehrte Vorzeichen.
Zusammen mit solchen Bändern werden Bänder beobachtet, die Übergängen mit einer Änderung des Trägheitsmoments entsprechen, jedoch mit. In diesem Fall sollten gemäß Formel (9) die Frequenzen der Linien davon abhängen und die Abstände zwischen den Linien ungleich werden. Jeder Streifen besteht aus einer Reihe von Linien, die sich zu einer Kante hin verdichten.
welches der Kopf des Streifens genannt wird. Für die Frequenz einer einzelnen im Band enthaltenen Spektrallinie gab Delander bereits 1885 eine empirische Formel der folgenden Form an:
wo ist eine ganze Zahl.
Delandres Formel folgt direkt aus den obigen Überlegungen. Delandres Formel lässt sich grafisch darstellen, wenn wir sie entlang einer Achse und entlang der anderen auftragen (Abb. 290).

Reis. 290. Grafische Darstellung der Delandre-Formel.
Unten sind die entsprechenden Linien zu sehen, die, wie wir sehen, einen typischen Streifen bilden. Da die Struktur des Molekülspektrums stark vom Trägheitsmoment des Moleküls abhängt, ist die Untersuchung von Molekülspektren eine der zuverlässigsten Methoden zur Bestimmung dieses Wertes. Die geringsten Veränderungen in der Struktur eines Moleküls können durch die Untersuchung seines Spektrums erkannt werden. Am interessantesten ist die Tatsache, dass Moleküle, die unterschiedliche Isotope (§ 86) desselben Elements enthalten, unterschiedliche Linien in ihrem Spektrum aufweisen sollten, die unterschiedlichen Massen dieser Isotope entsprechen. Dies folgt aus der Tatsache, dass die Massen der Atome sowohl die Frequenz ihrer Schwingungen im Molekül als auch dessen Trägheitsmoment bestimmen. Tatsächlich bestehen die Kupferchlorid-Bandlinien aus vier Komponenten, die vier Kombinationen der Kupferisotope 63 und 65 mit den Chlorisotopen 35 und 37 entsprechen:
Es wurden auch Linien entdeckt, die Molekülen entsprechen, die ein schweres Wasserstoffisotop enthalten, obwohl die Konzentration des Isotops in gewöhnlichem Wasserstoff gleich ist
Neben der Masse von Kernen beeinflussen auch andere Eigenschaften von Kernen die Struktur von Molekülspektren. Insbesondere die Rotationsmomente (Spins) der Kerne spielen eine sehr wichtige Rolle. Wenn in einem Molekül, das aus identischen Atomen besteht, die Rotationsmomente der Kerne gleich Null sind, fällt jede zweite Linie des Rotationsbandes aus. Dieser Effekt wird beispielsweise im Molekül beobachtet
Wenn die Rotationsmomente der Kerne ungleich Null sind, können sie einen Intensitätswechsel im Rotationsband verursachen, wobei sich schwache Linien mit starken abwechseln.)
Schließlich war es mithilfe radiospektroskopischer Methoden möglich, die Hyperfeinstruktur molekularer Spektren, die mit dem elektrischen Quadrupolmoment von Kernen verbunden sind, zu erkennen und genau zu messen.
Das elektrische Quadrupolmoment entsteht durch die Abweichung der Kernform von der Kugelform. Der Kern kann die Form eines länglichen oder abgeflachten Rotationsellipsoids haben. Ein solch geladenes Ellipsoid kann nicht mehr einfach durch eine Punktladung im Zentrum des Kerns ersetzt werden.

Reis. 291. Absorbierende Vorrichtung für „Atomuhren“: 1 – ein rechteckiger Wellenleiter mit einem Querschnitt einer Länge, der auf beiden Seiten durch gasdichte Schotte 7 verschlossen und mit Ammoniak bei niedrigem Druck gefüllt ist;
2 - Kristalldiode, die Harmonische der ihr zugeführten Hochfrequenzspannung erzeugt; 3 - Ausgangskristalldiode; 4 - Generator frequenzmodulierter Hochfrequenzspannung; 5 - Rohrleitung zur Vakuumpumpe und zum Ammoniakgasbehälter; 6 – Ausgabe an einen Impulsverstärker; 7 - Schotte; I - Kristalldioden-Stromanzeige; B - Vakuummeter.
Zusätzlich zur Coulomb-Kraft tritt im Kernfeld eine zusätzliche Kraft auf, die umgekehrt proportional zur vierten Potenz des Abstands und abhängig vom Winkel mit der Richtung der Symmetrieachse des Kerns ist. Das Auftreten zusätzlicher Kraft ist mit dem Vorhandensein eines Quadrupolmoments am Kern verbunden.
Zum ersten Mal wurde das Vorhandensein eines Quadrupolmoments in einem Kern durch konventionelle Spektroskopie anhand einiger Details der Hyperfeinstruktur von Atomlinien nachgewiesen. Diese Methoden ermöglichten es jedoch nicht, die Größe des Augenblicks genau zu bestimmen.
Bei der radiospektroskopischen Methode wird ein Wellenleiter mit dem zu untersuchenden molekularen Gas gefüllt und die Absorption von Radiowellen im Gas gemessen. Die Verwendung von Klystronen zur Erzeugung von Radiowellen ermöglicht es, Schwingungen mit einem hohen Grad an Monochromatizität zu erzeugen, die dann moduliert werden. Besonders detailliert wurde das Absorptionsspektrum von Ammoniak im Zentimeterwellenbereich untersucht. In diesem Spektrum wurde eine Hyperfeinstruktur entdeckt, die durch das Vorhandensein eines Zusammenhangs zwischen dem Quadrupolmoment des Kerns und dem elektrischen Feld des Moleküls selbst erklärt wird.
Der grundlegende Vorteil der Radiospektroskopie ist die niedrige Energie der Photonen, die den Radiofrequenzen entsprechen. Dank dessen kann die Absorption von Radiofrequenzen Übergänge zwischen extrem nahe beieinander liegenden Energieniveaus von Atomen und Molekülen erkennen. Neben nuklearen Effekten eignet sich die Radiospektroskopie-Methode sehr gut zur Bestimmung der elektrischen Dipolmomente des gesamten Moleküls durch den Stark-Effekt molekularer Linien in schwacher Elektrizität
Felder. In den letzten Jahren ist eine Vielzahl von Arbeiten erschienen, die sich der radiospektroskopischen Methode zur Untersuchung der Struktur verschiedenster Moleküle widmen. Die Absorption von Radiowellen in Ammoniak wurde zum Bau ultrapräziser „Atomuhren“ genutzt (Abb. 291).
Die Dauer des astronomischen Tages nimmt langsam zu und schwankt zudem innerhalb der Grenzen. Es ist wünschenswert, Uhren mit einem gleichmäßigeren Gang zu bauen. Eine „Atomuhr“ ist ein Quarzgenerator für Radiowellen, dessen Frequenz durch die Absorption der erzeugten Wellen in Ammoniak gesteuert wird. Bei einer Wellenlänge von 1,25 cm tritt Resonanz mit der Eigenfrequenz des Ammoniakmoleküls auf, was einer sehr scharfen Absorptionslinie entspricht. Die geringste Abweichung der Generatorwellenlänge von diesem Wert stört die Resonanz und führt zu einer starken Erhöhung der Transparenz des Gases für die Funkemission, die von den entsprechenden Geräten registriert wird und die Automatisierung aktiviert, die die Frequenz des Generators wiederherstellt. „Atomuhren“ haben sich bereits gleichmäßiger bewegt als die Erdrotation. Es wird davon ausgegangen, dass eine Genauigkeit in der Größenordnung von Bruchteilen eines Tages erreicht werden kann.

Untersuchungen molekularer Spektren ermöglichen die Bestimmung der zwischen Atomen in einem Molekül wirkenden Kräfte, der Dissoziationsenergie des Moleküls, seiner Geometrie, Kernabstände usw. , d.h. liefern umfassende Informationen über die Struktur und Eigenschaften des Moleküls.
Das Molekülspektrum bezeichnet im weitesten Sinne die Verteilung der Wahrscheinlichkeit von Übergängen zwischen einzelnen zwei Energieniveaus eines Moleküls (siehe Abb. 9) in Abhängigkeit von der Übergangsenergie. Da es im Folgenden um optische Spektren geht, muss jeder dieser Übergänge mit der Emission oder Absorption eines Photons mit Energie einhergehen
E n = hn = E 2 – E 1, 3.1
wobei E 2 und E 1 die Energien der Ebenen sind, zwischen denen der Übergang stattfindet.
Wenn Strahlung, die aus von Gasmolekülen emittierten Photonen besteht, durch ein Spektralgerät geleitet wird, erhält man das Emissionsspektrum des Moleküls, das aus einzelnen hellen (ggf. farbigen) Linien besteht. Darüber hinaus entspricht jede Zeile dem entsprechenden Übergang. Helligkeit und Position der Linie im Spektrum wiederum hängen von der Übergangswahrscheinlichkeit bzw. der Energie (Frequenz, Wellenlänge) des Photons ab.
Wenn dagegen Strahlung, die aus Photonen aller Wellenlängen besteht (kontinuierliches Spektrum), durch dieses Gas und dann durch ein Spektralgerät geleitet wird, erhält man ein Absorptionsspektrum. In diesem Fall besteht dieses Spektrum aus einer Reihe dunkler Linien vor dem Hintergrund eines hellen, kontinuierlichen Spektrums. Kontrast und Lage der Linie im Spektrum hängen dabei auch von der Übergangswahrscheinlichkeit und der Photonenenergie ab.
Aufgrund der komplexen Struktur der Energieniveaus des Moleküls (siehe Abb. 9) können alle Übergänge zwischen ihnen in separate Typen unterteilt werden, die dem Spektrum der Moleküle einen unterschiedlichen Charakter verleihen.
Ein Spektrum, das aus Linien besteht, die Übergängen zwischen Rotationsniveaus entsprechen (siehe Abb. 8), ohne die Schwingungs- und elektronischen Zustände des Moleküls zu ändern, wird als Rotationsspektrum des Moleküls bezeichnet. Da die Energie der Rotationsbewegung im Bereich von 10 -3 -10 -5 eV liegt, sollte die Frequenz der Linien in diesen Spektren im Mikrowellenbereich der Radiofrequenzen (ferner Infrarotbereich) liegen.
Ein Spektrum, das aus Linien besteht, die Übergängen zwischen Rotationsniveaus entsprechen, die zu verschiedenen Schwingungszuständen eines Moleküls im gleichen elektronischen Zustand gehören, wird als Schwingungs-Rotations- oder einfach Schwingungsspektrum eines Moleküls bezeichnet. Diese Spektren mit Schwingungsenergien von 10 -1 -10 -2 eV liegen im Infrarot-Frequenzbereich.
Schließlich wird ein Spektrum, das aus Linien besteht, die Übergängen zwischen Rotationsniveaus entsprechen, die zu verschiedenen elektronischen und Schwingungszuständen des Moleküls gehören, als Elektronen-Schwingungs-Rotations- oder einfach elektronisches Spektrum des Moleküls bezeichnet. Diese Spektren liegen im sichtbaren und ultravioletten Frequenzbereich, weil Die Energie der elektronischen Bewegung beträgt mehrere Elektronenvolt.
Da es sich bei der Emission (oder Absorption) eines Photons um einen elektromagnetischen Prozess handelt, ist seine notwendige Bedingung das Vorhandensein oder genauer gesagt eine Änderung des elektrischen Dipolmoments, das mit dem entsprechenden Quantenübergang im Molekül verbunden ist. Daraus folgt, dass Rotations- und Schwingungsspektren nur für Moleküle beobachtet werden können, die ein elektrisches Dipolmoment besitzen, also bestehend aus unterschiedlichen Atomen.
Molekülspektren, optische Emissions- und Absorptionsspektren sowie Raman-Streuung, Zugehörigkeit zu frei oder lose verbunden Moleküle. MS. haben eine komplexe Struktur. Typische M. s. - gestreift, werden sie bei der Emission und Absorption sowie bei der Raman-Streuung in Form einer Reihe mehr oder weniger schmaler Banden im ultravioletten, sichtbaren und nahen Infrarotbereich beobachtet, die sich bei ausreichendem Auflösungsvermögen der verwendeten Spektralinstrumente in a auflösen Reihe eng beieinander liegender Linien. Die spezifische Struktur von M. s. ist für verschiedene Moleküle unterschiedlich und wird im Allgemeinen komplexer, je mehr Atome im Molekül vorhanden sind. Bei sehr komplexen Molekülen bestehen die sichtbaren und ultravioletten Spektren aus wenigen breiten kontinuierlichen Banden; die Spektren solcher Moleküle sind einander ähnlich.
MS. entstehen wann Quantenübergänge zwischen Energieniveaus E' Und E'' Moleküle entsprechend dem Verhältnis
H n= E‘ - E‘’, (1)
Wo H n - Energie der emittierten absorbierten Energie Photon Frequenz n ( H -Plancksche Konstante ). Mit Raman-Streuung H n ist gleich der Differenz zwischen den Energien der einfallenden und gestreuten Photonen. MS. viel komplexer als Linienatomspektren, was durch die größere Komplexität der inneren Bewegungen in einem Molekül als in Atomen bestimmt wird. Zusammen mit der Bewegung von Elektronen relativ zu zwei oder mehr Kernen in Molekülen kommt es zu einer Schwingungsbewegung der Kerne (zusammen mit den sie umgebenden inneren Elektronen) um Gleichgewichtspositionen und einer Rotationsbewegung des Moleküls als Ganzes. Diese drei Bewegungsarten – elektronische, Vibrations- und Rotationsbewegung – entsprechen drei Arten von Energieniveaus und drei Arten von Spektren.
Nach der Quantenmechanik kann die Energie aller Bewegungsarten in einem Molekül nur bestimmte Werte annehmen, d. h. sie ist quantisiert. Gesamtenergie eines Moleküls E kann näherungsweise als Summe der quantisierten Energiewerte von drei Arten seiner Bewegung dargestellt werden:
E = E E-Mail + E zählen + E drehen (2)
Nach Größenordnung
Wo M ist die Masse des Elektrons und die Größe M hat die Größenordnung der Atomkerne in einem Molekül, d.h. m/M~ 10 -3 -10 -5, daher:
E E-Mail >> E zählen >> E drehen (4)
Gewöhnlich E el über mehrere ev(einige Hundert kJ/mol), E zählen ~ 10 -2 -10 -1 Vorabend Drehung ~ 10 -5 -10 -3 ev.
Gemäß (4) ist das System der Energieniveaus eines Moleküls durch eine Reihe weit voneinander entfernter elektronischer Niveaus (unterschiedliche Werte) gekennzeichnet E el bei E zählen = E Rotation = 0), Schwingungsniveaus, die viel näher beieinander liegen (unterschiedliche Werte). E bei einem gegebenen Wert zählen E Land E Rotation = 0) und noch dichter beieinander liegende Rotationsebenen (verschiedene Werte). E Rotation bei gegeben E el und E zählen).
Elektronische Energieniveaus ( E el in (2) entsprechen den Gleichgewichtskonfigurationen des Moleküls (im Fall eines zweiatomigen Moleküls, gekennzeichnet durch den Gleichgewichtswert). R 0 Kernabstand R. Jeder elektronische Zustand entspricht einer bestimmten Gleichgewichtskonfiguration und einem bestimmten Wert E el; der niedrigste Wert entspricht dem Grundenergieniveau.
Der Satz elektronischer Zustände eines Moleküls wird durch die Eigenschaften seiner Elektronenhülle bestimmt. Im Prinzip die Werte E el kann mit Methoden berechnet werden Quantenchemie, Allerdings kann dieses Problem nur mit Näherungsmethoden und für relativ einfache Moleküle gelöst werden. Die wichtigsten Informationen über die elektronischen Niveaus eines Moleküls (die Lage der elektronischen Energieniveaus und ihre Eigenschaften), die durch seine chemische Struktur bestimmt werden, werden durch die Untersuchung seiner Molekülstruktur gewonnen.
Ein sehr wichtiges Merkmal eines bestimmten elektronischen Energieniveaus ist der Wert Quantenzahl S, Charakterisierung des Absolutwerts des Gesamtspinmoments aller Elektronen des Moleküls. Chemisch stabile Moleküle haben normalerweise eine gerade Anzahl an Elektronen, und zwar für sie S= 0, 1, 2... (für die elektronische Hauptebene beträgt der typische Wert S= 0, und für aufgeregte - S= 0 und S= 1). Ebenen mit S= 0 heißen Singulett, mit S= 1 - Triplett (da die Wechselwirkung im Molekül zu deren Aufspaltung in c = 2 führt S+ 1 = 3 Unterebenen) . MIT freie Radikale haben in der Regel eine für sie ungerade Anzahl an Elektronen S= 1 / 2, 3 / 2, ... und der Wert ist sowohl für die Haupt- als auch die angeregte Ebene typisch S= 1 / 2 (Dublettebenen, die sich in c = 2 Unterebenen aufteilen).
Für Moleküle, deren Gleichgewichtskonfiguration symmetrisch ist, können die elektronischen Niveaus weiter klassifiziert werden. Im Fall von zweiatomigen und linearen dreiatomigen Molekülen mit einer Symmetrieachse (unendlicher Ordnung), die durch die Kerne aller Atome verläuft , Elektronische Niveaus werden durch die Werte der Quantenzahl l charakterisiert, die den Absolutwert der Projektion des gesamten Bahnimpulses aller Elektronen auf die Achse des Moleküls bestimmt. Ebenen mit l = 0, 1, 2, ... werden jeweils mit S, P, D... bezeichnet, und der Wert von c wird durch den Index oben links angezeigt (z. B. 3 S, 2 p, ...). Für Moleküle mit einem Symmetriezentrum, zum Beispiel CO 2 und C 6 H 6 , Alle elektronischen Ebenen sind in gerade und ungerade unterteilt und durch Indizes gekennzeichnet G Und u(abhängig davon, ob die Wellenfunktion bei Umkehrung im Symmetriezentrum ihr Vorzeichen behält oder ändert).
Schwingungsenergieniveaus (Werte E Anzahl) kann durch Quantisierung der Schwingungsbewegung ermittelt werden, die annähernd als harmonisch angesehen wird. Im einfachsten Fall eines zweiatomigen Moleküls (ein Schwingungsfreiheitsgrad, entsprechend einer Änderung des Kernabstands). R) wird es als harmonisch betrachtet Oszillator; seine Quantisierung ergibt gleichmäßig verteilte Energieniveaus:
E zählen = H n e (u +1/2), (5)
wobei n e die Grundfrequenz der harmonischen Schwingungen des Moleküls ist, u die Schwingungsquantenzahl ist und die Werte 0, 1, 2, ... annimmt. Für jeden elektronischen Zustand eines mehratomigen Moleküls bestehend aus N Atome ( N³ 3) und haben F Schwingungsfreiheitsgrade ( F = 3N- 5 und F = 3N- 6 für lineare bzw. nichtlineare Moleküle), stellt sich heraus F sogenannt Normalschwingungen mit Frequenzen n i ( ich = 1, 2, 3, ..., F) und ein komplexes System von Schwingungsniveaus:
![]()
Wo u i = 0, 1, 2, ... sind die entsprechenden Schwingungsquantenzahlen. Der Frequenzsatz der Normalschwingungen im elektronischen Grundzustand ist abhängig von seiner chemischen Struktur ein sehr wichtiges Merkmal eines Moleküls. Alle oder ein Teil der Atome des Moleküls nehmen an einer bestimmten Normalschwingung teil; Die Atome führen harmonische Schwingungen mit der gleichen Frequenz aus v i, aber mit unterschiedlichen Amplituden, die die Form der Schwingung bestimmen. Normalschwingungen werden entsprechend ihrer Form in Streckung (bei der sich die Längen der Bindungslinien ändern) und Biegung (bei der sich die Winkel zwischen chemischen Bindungen – Bindungswinkel – ändern) unterteilt. Die Anzahl der unterschiedlichen Schwingungsfrequenzen für Moleküle mit niedriger Symmetrie (ohne Symmetrieachsen mit einer höheren Ordnung als 2) beträgt 2, und alle Schwingungen sind nicht entartet, während es für symmetrischere Moleküle zweifach und dreifach entartete Schwingungen (Paare und Tripletts) gibt von Schwingungen, die in der Frequenz übereinstimmen). Zum Beispiel in einem nichtlinearen dreiatomigen Molekül H 2 O F= 3 und drei nicht entartete Schwingungen sind möglich (zwei Streckungen und eine Biegung). Das symmetrischere lineare dreiatomige CO 2 -Molekül hat F= 4 - zwei nicht entartete Schwingungen (Dehnung) und eine doppelt entartete Schwingung (Verformung). Für ein flaches hochsymmetrisches Molekül C 6 H 6 stellt sich heraus F= 30 - zehn nicht entartete und 10 doppelt entartete Schwingungen; Davon treten 14 Schwingungen in der Ebene des Moleküls auf (8 Streckungen und 6 Biegungen) und 6 Biegeschwingungen außerhalb der Ebene – senkrecht zu dieser Ebene. Das noch symmetrischere tetraedrische CH 4 -Molekül hat f = 9 - eine nicht entartete Schwingung (Streckung), eine zweifach entartete Schwingung (Verformung) und zwei dreifach entartete Schwingung (eine Streckung und eine Verformung).
Rotationsenergieniveaus können ermittelt werden, indem man die Rotationsbewegung eines Moleküls quantisiert und es mit Sicherheit als Festkörper behandelt Trägheitsmomente. Im einfachsten Fall eines zweiatomigen oder linearen mehratomigen Moleküls seine Rotationsenergie
![]()
Wo ICH ist das Trägheitsmoment des Moleküls relativ zu einer Achse senkrecht zur Achse des Moleküls und M- Rotationsimpuls. Gemäß den Quantisierungsregeln gilt
![]()
Wo ist die Rotationsquantenzahl? J= 0, 1, 2, ... und daher für E Rotation erhalten:
wobei die Rotationskonstante den Maßstab der Abstände zwischen den Energieniveaus bestimmt, der mit zunehmender Kernmasse und Kernabständen abnimmt.
Verschiedene Arten von M. s. entstehen bei verschiedenen Arten von Übergängen zwischen Energieniveaus von Molekülen. Nach (1) und (2)
D E = E‘ - E'' = D E el + D E zählen + D E drehen, (8)
wo ändert sich D E el, D E Graf und D E Die Rotation elektronischer, Schwingungs- und Rotationsenergien erfüllt die Bedingung:
D E el >> D E zählen >> D E drehen (9)
[Abstände zwischen Ebenen sind von der gleichen Größenordnung wie die Energien selbst E el, E ol und E Rotation, erfüllt Bedingung (4)].
Bei D E el ¹ 0, Elektronenmikroskopie wird erhalten, die im sichtbaren und ultravioletten (UV) Bereich beobachtet wird. Normalerweise bei D E el ¹ 0 gleichzeitig D E Nummer 0 und D E Drehung ¹ 0; anders D E zählen für ein gegebenes D E el entsprechen verschiedenen Schwingungsbändern und unterschiedlichen D E Drehung bei gegebenem D E el und d E zählen - einzelne Rotationslinien, in die dieser Streifen zerfällt; Es entsteht eine charakteristische Streifenstruktur.
![]()
Rotationsaufspaltung der Elektronenschwingungsbande 3805 des N 2 -Moleküls
Eine Reihe von Streifen mit einem bestimmten D E el (entspricht einem rein elektronischen Übergang mit einer Frequenz v el = D E Email/ H) wird als Streifensystem bezeichnet; Abhängig von den relativen Übergangswahrscheinlichkeiten, die mit quantenmechanischen Methoden näherungsweise berechnet werden können, weisen einzelne Bänder unterschiedliche Intensitäten auf. Bei komplexen Molekülen verschmelzen die Bänder eines Systems, die einem bestimmten elektronischen Übergang entsprechen, normalerweise zu einem breiten kontinuierlichen Band; mehrere solcher breiten Bänder können einander überlappen. Charakteristische diskrete elektronische Spektren, die in gefrorenen Lösungen organischer Verbindungen beobachtet werden . Elektronische (genauer gesagt elektronische Schwingungs-Rotations-)Spektren werden experimentell mit Spektrographen und Spektrometern mit Glasoptik (für den sichtbaren Bereich) und Quarzoptik (für den UV-Bereich) untersucht, bei denen Prismen oder Beugungsgitter verwendet werden, um Licht in a zu zerlegen Spektrum .
Bei D E el = 0 und D E Zählung ¹ 0, es werden oszillatorische Magnetresonanzen erhalten, die im Nahbereich (bis zu mehreren) beobachtet werden µm) und in der Mitte (bis zu mehreren Zehnern). µm) Infrarot (IR)-Bereich, normalerweise bei der Absorption sowie bei der Raman-Streuung von Licht. In der Regel gleichzeitig D E Drehung ¹ 0 und bei einem gegebenen E Das Ergebnis ist ein Schwingungsband, das in einzelne Rotationslinien zerfällt. Am intensivsten sind sie bei oszillierenden M. s. Streifen entsprechend D u = u’ - u'' = 1 (für mehratomige Moleküle - D u ich = u ich' - u i ''= 1 bei D u k = u k’ – u k '' = 0, wo k¹i).
Für rein harmonische Schwingungen sind diese Auswahlregeln, das Verbot anderer Übergänge wird strikt durchgeführt; für anharmonische Schwingungen treten Bänder auf, für die D u> 1 (Obertöne); Ihre Intensität ist normalerweise gering und nimmt mit zunehmendem D ab u.
Schwingungsspektren (genauer gesagt Schwingungs-Rotations-Spektren) werden experimentell im IR-Bereich in der Absorption mit IR-Spektrometern mit für IR-Strahlung transparenten Prismen oder mit Beugungsgittern sowie Fourier-Spektrometern und in der Raman-Streuung mit Spektrographen mit hoher Apertur untersucht ( für die sichtbaren Bereich) mittels Laseranregung.
Bei D E el = 0 und D E count = 0 erhält man rein rotatorische Magnetsysteme, bestehend aus einzelnen Linien. Sie werden bei Absorption aus einer Entfernung (Hunderte von) beobachtet µm)IR-Bereich und insbesondere im Mikrowellenbereich sowie in Raman-Spektren. Bei zweiatomigen und linearen mehratomigen Molekülen (sowie bei ziemlich symmetrischen nichtlinearen mehratomigen Molekülen) haben diese Linien den gleichen Abstand (auf der Frequenzskala) voneinander mit Intervallen Dn = 2 B in Absorptionsspektren und Dn = 4 B in Raman-Spektren.
Reine Rotationsspektren werden in der Absorption im fernen IR-Bereich mit IR-Spektrometern mit speziellen Beugungsgittern (Echelettes) und Fourier-Spektrometern untersucht, im Mikrowellenbereich mit Mikrowellen-(Mikrowellen-)Spektrometern , sowie in der Raman-Streuung unter Verwendung von Spektrographen mit hoher Apertur.
Methoden der molekularen Spektroskopie, die auf der Untersuchung von Mikroorganismen basieren, ermöglichen die Lösung verschiedener Probleme in der Chemie, Biologie und anderen Wissenschaften (z. B. Bestimmung der Zusammensetzung von Erdölprodukten, Polymersubstanzen usw.). In Chemie nach MS. Studieren Sie die Struktur von Molekülen. Elektronische M. s. ermöglichen es, Informationen über die elektronischen Hüllen von Molekülen zu erhalten, angeregte Niveaus und ihre Eigenschaften zu bestimmen und die Dissoziationsenergien von Molekülen zu ermitteln (durch die Konvergenz der Schwingungsniveaus eines Moleküls an den Dissoziationsgrenzen). Studium der oszillierenden M. s. ermöglicht es Ihnen, die charakteristischen Schwingungsfrequenzen zu finden, die bestimmten Arten chemischer Bindungen im Molekül (z. B. einfache Doppel- und Dreifach-C-C-Bindungen, C-H-, N-H-, O-H-Bindungen für organische Moleküle) und verschiedenen Atomgruppen (z. B. CH 2) entsprechen , CH 3 , NH 2), bestimmen die räumliche Struktur von Molekülen, unterscheiden zwischen cis- und trans-Isomeren. Hierzu werden sowohl Infrarot-Absorptionsspektren (IR) als auch Raman-Spektren (RSS) verwendet. Besonders verbreitet ist die IR-Methode als eine der effektivsten optischen Methoden zur Untersuchung der Struktur von Molekülen. In Kombination mit der SKR-Methode liefert es die umfassendsten Informationen. Die Untersuchung rotatorischer Magnetresonanzen sowie der Rotationsstruktur elektronischer und Schwingungsspektren ermöglicht die Nutzung experimentell ermittelter Werte der Trägheitsmomente von Molekülen [die aus den Werten der Rotationskonstanten gewonnen werden, vgl (7)] mit großer Genauigkeit (für einfachere Moleküle, zum Beispiel H 2 O) Parameter der Gleichgewichtskonfiguration des Moleküls zu finden – Bindungslängen und Bindungswinkel. Um die Anzahl der ermittelten Parameter zu erhöhen, werden die Spektren von Isotopenmolekülen (insbesondere bei denen Wasserstoff durch Deuterium ersetzt wird) mit den gleichen Parametern der Gleichgewichtskonfiguration, aber unterschiedlichen Trägheitsmomenten untersucht.
Als Beispiel für die Verwendung von M. s. Um die chemische Struktur von Molekülen zu bestimmen, betrachten Sie das Benzolmolekül C 6 H 6 . Studiert ihren M. s. bestätigt die Richtigkeit des Modells, wonach das Molekül flach ist und alle 6 C-C-Bindungen im Benzolring äquivalent sind und ein regelmäßiges Sechseck mit einer Symmetrieachse sechster Ordnung bilden, die durch das Symmetriezentrum des Moleküls senkrecht zu seinem verläuft Flugzeug. Elektronische M. s. Die Absorptionsbande C 6 H 6 besteht aus mehreren Bandensystemen, die Übergängen vom geraden Grund-Singulett-Niveau zu angeregten ungeraden Niveaus entsprechen, von denen das erste ein Triplett und die höheren ein Singulett-Niveau sind. Das Streifensystem ist im Bereich von 1840 am intensivsten ( E 5 - E 1 = 7,0 ev), das Bandsystem ist im Bereich von 3400 am schwächsten ( E 2 - E 1 = 3,8ev), entsprechend dem Singulett-Triplett-Übergang, der durch die Näherungsauswahlregeln für den Gesamtspin verboten ist. Übergänge entsprechen der Anregung des sogenannten. p-Elektronen sind im gesamten Benzolring delokalisiert ; Das aus elektronischen Molekülspektren erhaltene Niveaudiagramm stimmt mit näherungsweise quantenmechanischen Berechnungen überein. Oszillatorischer M. s. C 6 H 6 entsprechen dem Vorhandensein eines Symmetriezentrums im Molekül – Schwingungsfrequenzen, die im IRS erscheinen (aktiv), fehlen im SRS (inaktiv) und umgekehrt (das sogenannte alternative Verbot). Von den 20 normalen Schwingungen von C 6 H 6 4 sind im ICS aktiv und 7 im SCR aktiv, die restlichen 11 sind sowohl im ICS als auch im SCR inaktiv. Gemessene Frequenzwerte (in cm -1): 673, 1038, 1486, 3080 (in ICS) und 607, 850, 992, 1178, 1596, 3047, 3062 (in TFR). Die Frequenzen 673 und 850 entsprechen nichtebenen Schwingungen, alle anderen Frequenzen entsprechen ebenen Schwingungen. Besonders charakteristisch für planare Schwingungen sind die Frequenz 992 (entsprechend der Streckschwingung von C-C-Bindungen, bestehend aus periodischer Kompression und Streckung des Benzolrings), die Frequenzen 3062 und 3080 (entsprechend den Streckschwingungen von C-H-Bindungen) und die Frequenz 607 (entsprechend zur Biegeschwingung des Benzolrings). Die beobachteten Schwingungsspektren von C 6 H 6 (und ähnliche Schwingungsspektren von C 6 D 6) stimmen sehr gut mit theoretischen Berechnungen überein, die eine vollständige Interpretation dieser Spektren und die Ermittlung der Formen aller Normalschwingungen ermöglichten.
Auf die gleiche Weise können Sie M. s. verwenden. Bestimmen Sie die Struktur verschiedener Klassen organischer und anorganischer Moleküle, bis hin zu sehr komplexen, wie z. B. Polymermolekülen.
Vorlesung 12. Kernphysik. Der Aufbau des Atomkerns.
Kern- Dies ist der zentrale massive Teil des Atoms, um den sich Elektronen in Quantenbahnen drehen. Die Masse des Atomkerns ist etwa 4·10 3 mal größer als die Masse aller im Atom enthaltenen Elektronen. Die Kernelgröße ist sehr klein (10 -12 -10 -13). cm), was ungefähr 10 5 mal kleiner ist als der Durchmesser des gesamten Atoms. Die elektrische Ladung ist positiv und entspricht im Absolutwert der Summe der Ladungen der Atomelektronen (da das Atom als Ganzes elektrisch neutral ist).
Der Kern wurde von E. Rutherford (1911) in Experimenten zur Streuung von Alphateilchen beim Durchgang durch Materie entdeckt. Nachdem Rutherford entdeckt hatte, dass a-Teilchen häufiger als erwartet in großen Winkeln gestreut werden, schlug er vor, dass die positive Ladung des Atoms in einem kleinen Kern konzentriert ist (davor herrschten die Ideen von J. Thomson vor, wonach die positive Ladung von Das Atom wurde als gleichmäßig über sein gesamtes Volumen verteilt betrachtet. Rutherfords Idee wurde von seinen Zeitgenossen nicht sofort akzeptiert (das Haupthindernis war der Glaube an den unvermeidlichen Fall atomarer Elektronen auf den Kern aufgrund des Energieverlusts durch elektromagnetische Strahlung bei der Bewegung in der Umlaufbahn um den Kern). Maßgeblichen Anteil an ihrer Anerkennung hatte das berühmte Werk von N. Bohr (1913), das den Grundstein für die Quantentheorie des Atoms legte. Bohr postulierte die Stabilität von Umlaufbahnen als Ausgangsprinzip der Quantisierung der Bewegung atomarer Elektronen und leitete daraus dann die Gesetze der optischen Linienspektren ab, die umfangreiches empirisches Material erklärten (Balmer-Reihe usw.). Etwas später (Ende 1913) zeigte Rutherfords Schüler G. Moseley experimentell, dass sich die kurzwellige Grenze der Linienröntgenspektren von Atomen verschiebt, wenn sich die Ordnungszahl Z eines Elements im Periodensystem der Elemente ändert entspricht Bohrs Theorie, wenn wir annehmen, dass die elektrische Ladung des Kerns (in Einheiten der Elektronenladung) gleich Z ist. Diese Entdeckung durchbrach die Barriere des Misstrauens völlig: Ein neues physikalisches Objekt – der Kern – erwies sich als fest verbunden mit einer ganzen Reihe scheinbar heterogener Phänomene, die nun eine einheitliche und physikalisch transparente Erklärung erhalten haben. Nach Moseleys Arbeit wurde die Tatsache der Existenz des Atomkerns endgültig in der Physik nachgewiesen.
Kernelzusammensetzung. Zum Zeitpunkt der Entdeckung des Kerns waren nur zwei Elementarteilchen bekannt – das Proton und das Elektron. Dementsprechend wurde es als wahrscheinlich angesehen, dass der Kern aus ihnen besteht. Allerdings Ende der 20er Jahre. 20. Jahrhundert Die Proton-Elektron-Hypothese stieß auf eine ernsthafte Schwierigkeit, die als „Stickstoffkatastrophe“ bezeichnet wurde: Gemäß der Proton-Elektron-Hypothese sollte der Stickstoffkern 21 Teilchen (14 Protonen und 7 Elektronen) enthalten, von denen jedes einen Spin von 1/2 hatte . Der Spin des Stickstoffkerns hätte halbzahlig sein sollen, aber nach den Daten zur Messung optischer Molekülspektren stellte sich heraus, dass der Spin gleich 1 war.
Die Zusammensetzung des Kerns wurde nach der Entdeckung von J. Chadwick (1932) geklärt. Neutron. Die Masse des Neutrons liegt, wie sich aus Chadwicks ersten Experimenten herausstellte, nahe an der Masse des Protons und der Spin beträgt 1/2 (später festgestellt). Die Idee, dass der Kern aus Protonen und Neutronen besteht, wurde erstmals von D. D. Ivanenko (1932) in gedruckter Form zum Ausdruck gebracht und unmittelbar danach von W. Heisenberg (1932) entwickelt. Die Annahme über die Protonen-Neutronen-Zusammensetzung des Kerns wurde später experimentell vollständig bestätigt. In der modernen Kernphysik werden Proton (p) und Neutron (n) häufig unter der gemeinsamen Bezeichnung Nukleon zusammengefasst. Die Gesamtzahl der Nukleonen in einem Kern wird Massenzahl genannt A, die Anzahl der Protonen ist gleich der Ladung des Kerns Z (in Einheiten der Elektronenladung), der Anzahl der Neutronen N = A - Z. U Isotope gleiches Z, aber anders A Und N, die Kerne haben die gleichen Isobaren A und verschiedene Z und N.
Im Zusammenhang mit der Entdeckung neuer Teilchen, die schwerer als Nukleonen sind, den sogenannten. Nukleonenisobaren stellten sich heraus, dass sie auch Teil des Kerns sein sollten (intranukleare Nukleonen, die miteinander kollidieren, können sich in Nukleonenisobaren verwandeln). Im einfachsten Kernel - Deuteron , bestehend aus einem Proton und einem Neutron, sollten Nukleonen in etwa 1 % der Fälle in Form von Nukleonenisobaren verbleiben. Eine Reihe beobachteter Phänomene zeugen für die Existenz solcher isobaren Zustände in Kernen. Zusätzlich zu Nukleonen und Nukleonenisobaren, in Kernen periodisch für kurze Zeit (10 -23 -10 -24). Sek) erscheinen Mesonen , einschließlich der leichtesten von ihnen - p-Mesonen. Die Wechselwirkung von Nukleonen beruht auf mehreren Vorgängen der Emission eines Mesons durch eines der Nukleonen und seiner Absorption durch ein anderes. Emerging dh. Austauschmesonenströme beeinflussen insbesondere die elektromagnetischen Eigenschaften von Kernen. Die deutlichste Manifestation von Mesonenaustauschströmen wurde in der Reaktion der Deuteronspaltung durch hochenergetische Elektronen und g-Quanten gefunden.
Wechselwirkung von Nukleonen. Die Kräfte, die Nukleonen im Kern halten, werden genannt nuklear . Dies sind die stärksten Wechselwirkungen, die in der Physik bekannt sind. Die Kernkräfte, die zwischen zwei Nukleonen in einem Kern wirken, sind um eine Größenordnung hundertmal stärker als die elektrostatische Wechselwirkung zwischen Protonen. Eine wichtige Eigenschaft der Nuklearstreitkräfte ist ihre. Unabhängigkeit vom Ladungszustand der Nukleonen: Die Kernwechselwirkungen zweier Protonen, zweier Neutronen oder eines Neutrons und eines Protons sind gleich, wenn die relativen Bewegungszustände dieser Teilchenpaare gleich sind. Die Größe der Kernkräfte hängt vom Abstand zwischen den Nukleonen, von der gegenseitigen Ausrichtung ihrer Spins, von der Ausrichtung der Spins relativ zum Bahndrehimpuls und vom Radiusvektor ab, der von einem Teilchen zum anderen gezogen wird. Kernkräfte zeichnen sich durch einen bestimmten Wirkungsbereich aus: Das Potenzial dieser Kräfte nimmt mit der Entfernung ab R zwischen Teilchen schneller als R-2, und die Kräfte selbst sind schneller als R-3. Aus der Betrachtung der physikalischen Natur der Kernkräfte folgt, dass sie mit der Entfernung exponentiell abnehmen sollten. Der Wirkungsradius nuklearer Kräfte wird durch die sogenannte bestimmt. Compton-Wellenlänge r 0 Mesonen, die während der Wechselwirkung zwischen Nukleonen ausgetauscht werden:
hier ist m die Mesonenmasse, das Plancksche Wirkungsquantum, Mit- Lichtgeschwindigkeit im Vakuum. Die durch den Austausch von p-Mesonen verursachten Kräfte haben den größten Wirkungsradius. Für sie ist r 0 = 1,41 F (1 f = 10 -13 cm). Die Abstände zwischen den Nukleonen in Kernen liegen genau in dieser Größenordnung, aber auch der Austausch schwererer Mesonen (m-, r-, w-Mesonen usw.) trägt zu Kernkräften bei. Die genaue Abhängigkeit der Kernkräfte zwischen zwei Nukleonen vom Abstand und dem Beitrag der Kernkräfte durch den Austausch von Mesonen unterschiedlicher Art ist nicht sicher geklärt. In Mehrkernkernen sind Kräfte möglich, die nicht auf die Wechselwirkung nur von Nukleonenpaaren reduziert werden können. Die Rolle dieser sogenannten Vielteilchenkräfte in der Struktur von Kernen sind weiterhin unklar.
Kernelgrößen hängen von der Anzahl der darin enthaltenen Nukleonen ab. Die durchschnittliche Dichte der Anzahl p der Nukleonen in einem Kern (ihre Anzahl pro Volumeneinheit) ist für alle Multinukleonenkerne (A > 0) nahezu gleich. Das bedeutet, dass das Volumen des Kerns proportional zur Anzahl der Nukleonen ist A und seine lineare Größe ~A 1/3. Effektiver Kernradius R wird durch die Beziehung bestimmt:
R = ein A 1/3 , (2)
Wo ist die Konstante? A nahe bei Hz, unterscheidet sich aber davon und hängt davon ab, an welchen physikalischen Phänomenen es gemessen wird R. Beim sogenannten Ladungsradius des Kerns, gemessen durch die Streuung von Elektronen an Kernen oder durch die Lage von Energieniveaus m- Mesoatome : a = 1,12 F. Aus Interaktionsprozessen ermittelter Wirkradius Hadronen (Nukleonen, Mesonen, a-Teilchen usw.) mit Kernen, die etwas größer als die Ladung sind: ab 1.2 F bis 1,4 F.
Die Dichte der Kernmaterie ist im Vergleich zur Dichte gewöhnlicher Substanzen fantastisch hoch: Sie beträgt etwa 10 14 G/cm 3. Im Kern ist r im zentralen Teil nahezu konstant und nimmt zur Peripherie hin exponentiell ab. Für eine ungefähre Beschreibung empirischer Daten wird manchmal die folgende Abhängigkeit von r vom Abstand r vom Mittelpunkt des Kerns akzeptiert:
![]() .
.
Effektiver Kernradius R gleich R 0 + b. Der Wert b charakterisiert die Unschärfe der Kerngrenze; er ist für alle Kerne nahezu gleich (» 0,5). F). Der Parameter r 0 ist die doppelte Dichte an der „Grenze“ des Kerns, bestimmt aus der Normalisierungsbedingung (Gleichheit des Volumenintegrals von p zur Anzahl der Nukleonen). A). Aus (2) folgt, dass die Größe der Kerne in der Größenordnung von 10 bis 13 variiert cm bis 10-12 cm für schwere Kerne (Atomgröße ~ 10 -8 cm). Allerdings beschreibt Formel (2) die Zunahme der Längenabmessungen von Kernen mit zunehmender Nukleonenzahl nur grob, mit deutlicher Zunahme A. Die Größenänderung des Kerns bei Zugabe von einem oder zwei Nukleonen hängt von den Details der Struktur des Kerns ab und kann unregelmäßig sein. Insbesondere (wie Messungen der Isotopenverschiebung atomarer Energieniveaus zeigen) verringert sich manchmal sogar der Radius des Kerns, wenn zwei Neutronen hinzugefügt werden.
MOLEKULARSPEKTRAEN, elektromagnetische Emissions- und Absorptionsspektren. Strahlung und Kombination Streuung des Lichts freier oder schwach gebundener Moleküle. Sie sehen aus wie eine Reihe von Bändern (Linien) im Röntgen-, UV-, sichtbaren, IR- und Radiowellenbereich (einschließlich Mikrowellen) des Spektrums. Die Lage der Banden (Linien) in den Emissionsspektren (Emissions-Molekülspektren) und der Absorption (Absorptions-Molekülspektren) wird durch Frequenzen v (Wellenlängen l = c/v, wobei c die Lichtgeschwindigkeit ist) und Wellenzahlen = 1 charakterisiert /l; sie wird durch die Differenz der Energien E" und E bestimmt: jene Zustände des Moleküls, zwischen denen ein Quantenübergang stattfindet:
![]()
(h-Planck-Konstante). Mit Kombination Bei der Streuung ist der Wert hv gleich der Differenz der Energien der einfallenden und gestreuten Photonen. Die Intensität der Bänder (Linien) hängt von der Anzahl (Konzentration) der Moleküle einer bestimmten Art, der Besetzung der Energieniveaus E" und E: und der Wahrscheinlichkeit des entsprechenden Übergangs ab.
Die Wahrscheinlichkeit von Übergängen mit Emission oder Absorption von Strahlung wird hauptsächlich durch das Quadrat des elektrischen Matrixelements bestimmt. Übergangsdipolmoment, und bei genauerer Betrachtung - durch die Quadrate der magnetischen Matrixelemente. und elektrisch Quadrupolmomente des Moleküls (siehe Quantenübergänge). Mit Kombination Bei der Lichtstreuung hängt die Übergangswahrscheinlichkeit mit dem Matrixelement des induzierten Übergangsdipolmoments des Moleküls zusammen, d. h. mit dem Matrixelement der Polarisierbarkeit des Moleküls.
Bedingungen sagen. Systeme, deren Übergänge in Form bestimmter Molekülspektren auftreten, sind unterschiedlicher Natur und unterscheiden sich stark in der Energie. Die Energieniveaus bestimmter Typen liegen weit voneinander entfernt, sodass das Molekül bei Übergängen hochfrequente Strahlung absorbiert oder aussendet. Der Abstand zwischen Ebenen anderer Art ist gering und in einigen Fällen mangels externer. die Feldebenen verschmelzen (entarten). Bei kleinen Energieunterschieden werden Übergänge im Niederfrequenzbereich beobachtet. Beispielsweise haben die Atomkerne bestimmter Elemente ihre eigenen. Mag. Drehmoment und Elektrik Quadrupolmoment, das mit dem Spin verbunden ist. Elektronen haben auch eine magnetische Wirkung Moment, der mit ihrem Spin verbunden ist. In Abwesenheit von externen magnetische Orientierungsfelder Momente sind willkürlich, d.h. sie sind nicht quantisiert und die entsprechenden Energien. Staaten sind degeneriert. Bei der externen Anwendung Dauermagnet Feld, die Entartung wird aufgehoben und Übergänge zwischen Energieniveaus sind möglich, die im Hochfrequenzbereich des Spektrums beobachtet werden. So entstehen NMR- und EPR-Spektren (siehe Kernspinresonanz, Elektronenparamagnetische Resonanz).
Kinetische Verteilung Energien der von Mol emittierten Elektronen. Systeme erzeugen durch Bestrahlung mit Röntgenstrahlung oder harter UV-Strahlung RöntgenstrahlungSpektroskopie und Photoelektronenspektroskopie. Zusätzlich Prozesse im Pier System, verursacht durch die anfängliche Anregung, führen zum Auftreten anderer Spektren. Auger-Spektren entstehen also durch Relaxation. Elektroneneinfang von außen Muscheln von k.-l. Atom pro freiem Inneren Schale, und die freigesetzte Energie wandelt sich um. in der Kinetik Energie eines anderen Elektrons ext. von einem Atom emittierte Hülle. In diesem Fall findet ein Quantenübergang von einem bestimmten Zustand eines neutralen Moleküls in einen Zustand eines Mols statt. Ion (siehe Auger-Spektroskopie).
Traditionell werden nur mit optischen Spektren verbundene Spektren als eigentliche Molekülspektren klassifiziert. Übergänge zwischen elektronisch-schwingungsrotierenden Energieniveaus eines Moleküls, die mit drei Grundelementen verbunden sind. Arten von Energie Ebenen des Moleküls – elektronisches E el, Schwingungs-E-Zählung und Rotations-E bp, entsprechend drei Arten von internen. Bewegung in einem Molekül. Die Energie der Gleichgewichtskonfiguration eines Moleküls in einem bestimmten elektronischen Zustand wird als Aal angenommen. Die Menge möglicher elektronischer Zustände eines Moleküls wird durch die Eigenschaften seiner elektronischen Hülle und Symmetrie bestimmt. Schwingung Die Bewegungen der Kerne in einem Molekül relativ zu ihrer Gleichgewichtsposition in jedem elektronischen Zustand werden für mehrere Schwingungen quantisiert. Freiheitsgrade entsteht ein komplexes Schwingungssystem. Energieniveaus E zählen. Durch Rotation ist die Rotation des Moleküls als Ganzes als starres System verbundener Kerne gekennzeichnet. Moment der Bewegungsmenge, die quantisiert wird und eine Drehung bildet. Zustände (Rotationsenergieniveaus) E Zeit. Typischerweise liegt die Energie elektronischer Übergänge in der Größenordnung von mehreren. eV, Vibration - 10 -2 ... 10 -1 eV, Rotation - 10 -5 ... 10 -3 eV.
Abhängig von den Energieniveaus treten Übergänge mit Emission, Absorption oder Kombinationen auf. elektromagnetische Streuung Strahlung - elektronisch, Schwingung. oder rotatorisch, es gibt elektronische Schwingungen. und Rotationsmolekülspektren. Die Artikel Elektronische Spektren, Schwingungsspektren, Rotationsspektren geben Auskunft über die entsprechenden Zustände von Molekülen, Auswahlregeln für Quantenübergänge, Mol. Spektroskopie sowie welche Eigenschaften von Molekülen genutzt werden können. aus Molekülspektren gewonnen: Eigenschaften und Symmetrie elektronischer Zustände, Schwingungen. Konstanten, Dissoziationsenergie, Symmetrie des Moleküls, Rotation. Konstanten, Trägheitsmomente, Geom. Parameter, elektrisch Dipolmomente, Strukturdaten und interne Kraftfelder usw. Elektronische Absorptions- und Lumineszenzspektren im sichtbaren und UV-Bereich geben Aufschluss über die Verteilung