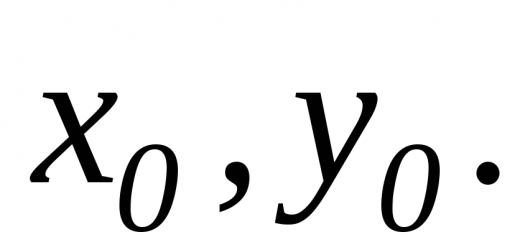Conférence n°6
Énergie moléculaire
Atome On appelle la plus petite particule d'un élément chimique qui a ses propriétés chimiques.
Un atome est constitué d'un noyau chargé positivement et d'électrons se déplaçant dans son champ. La charge du noyau est égale à la charge de tous les électrons. Ion d'un atome donné est une particule chargée électriquement formée lorsque les atomes perdent ou gagnent des électrons.
Molécule est la plus petite particule d'une substance homogène qui possède ses propriétés chimiques de base.
Les molécules sont constituées d'atomes identiques ou différents reliés entre eux par des liaisons chimiques interatomiques.
Afin de comprendre les raisons pour lesquelles des atomes électriquement neutres peuvent former une molécule stable, nous nous limiterons à considérer les molécules diatomiques les plus simples, constituées de deux atomes identiques ou différents.
Les forces qui maintiennent un atome dans une molécule sont provoquées par l’interaction d’électrons externes. Lorsque les atomes se combinent pour former une molécule, les électrons des couches internes restent dans leur état antérieur.
Si les atomes sont très éloignés les uns des autres, ils n’interagissent pas entre eux. À mesure que les atomes se rapprochent, les forces de leur attraction mutuelle augmentent. À des distances comparables à la taille des atomes, des forces de répulsion mutuelle apparaissent, qui ne permettent pas aux électrons d'un atome de pénétrer trop profondément dans les couches électroniques d'un autre atome.
Les forces répulsives sont plus « à courte portée » que les forces attractives. Cela signifie qu’à mesure que la distance entre les atomes augmente, les forces répulsives diminuent plus rapidement que les forces attractives.
Le graphique de la force attractive, de la force répulsive et de la force d'interaction résultante entre les atomes en fonction de la distance ressemble à ceci :
L'énergie d'interaction des électrons dans une molécule est déterminée par la disposition mutuelle des noyaux atomiques et est fonction de la distance, c'est-à-dire
L'énergie totale de la molécule entière comprend également l'énergie cinétique des noyaux en mouvement.
Ainsi,
![]() .
.
Cela signifie qu’il s’agit de l’énergie potentielle d’interaction entre les noyaux.
Représente alors la force d’interaction entre les atomes dans une molécule diatomique.
En conséquence, le graphique de la dépendance de l'énergie potentielle d'interaction des atomes dans une molécule sur la distance entre les atomes a la forme :

La distance interatomique d'équilibre dans une molécule est appelée longueur de connexion. La quantité D s'appelle énergie de dissociation moléculaire ou énergie de liaison. Il est numériquement égal au travail qui doit être effectué pour briser les liaisons chimiques des atomes en molécules et les éliminer au-delà de l'action des forces interatomiques. L'énergie de dissociation est égale à l'énergie libérée lors de la formation de la molécule, mais de signe opposé. L'énergie de dissociation est négative et l'énergie libérée lors de la formation d'une molécule est positive.
L'énergie d'une molécule dépend de la nature du mouvement des noyaux. Ce mouvement peut être divisé en translation, rotation et oscillation. À de petites distances entre les atomes d'une molécule et un volume suffisamment grand du récipient fourni aux molécules, énergie vers l'avant a un spectre continu et sa valeur moyenne est égale à , c'est-à-dire.
Énergie de rotation a un spectre discret et peut prendre des valeurs
![]() ,
,
où I est le nombre quantique de rotation ;
J est le moment d'inertie de la molécule.
Énergie du mouvement vibratoire possède également un spectre discret et peut prendre des valeurs
![]() ,
,
où est le nombre quantique vibrationnel ;
– fréquence propre de ce type d'oscillation.
Lorsque le niveau vibratoire le plus bas n’a aucune énergie
 L'énergie du mouvement de rotation et de translation correspond à la forme cinétique de l'énergie, l'énergie du mouvement oscillatoire correspond à la forme potentielle. Par conséquent, les étapes énergétiques du mouvement vibrationnel d’une molécule diatomique peuvent être représentées sur un graphique.
L'énergie du mouvement de rotation et de translation correspond à la forme cinétique de l'énergie, l'énergie du mouvement oscillatoire correspond à la forme potentielle. Par conséquent, les étapes énergétiques du mouvement vibrationnel d’une molécule diatomique peuvent être représentées sur un graphique.
Les étapes énergétiques du mouvement de rotation d'une molécule diatomique sont situées de la même manière, seule la distance entre elles est beaucoup plus petite que celle des mêmes étapes du mouvement vibratoire.
Principaux types de liaisons interatomiques
Il existe deux types de liaisons atomiques : ionique (ou hétéropolaire) et covalente (ou homéopolaire).
Liaison ionique se produit dans les cas où les électrons d'une molécule sont disposés de telle manière qu'un excès se forme près de l'un des noyaux et un déficit près de l'autre. Ainsi, la molécule semble être constituée de deux ions de signes opposés, attirés l'un vers l'autre. Des exemples de molécules avec des liaisons ioniques sont NaCl, KCl, RbF, CsJ etc. formé en combinant des atomes d'éléments je-Oh et VII-ème groupe du système périodique de Mendeleïev. Dans ce cas, un atome qui s'est ajouté un ou plusieurs électrons acquiert une charge négative et devient un ion négatif, et un atome qui donne le nombre correspondant d'électrons se transforme en un ion positif. La somme totale des charges positives et négatives des ions est nulle. Les molécules ioniques sont donc électriquement neutres. Les forces qui assurent la stabilité de la molécule sont de nature électrique.
Pour qu'une liaison ionique ait lieu, il faut que l'énergie d'élimination des électrons, c'est-à-dire le travail de création d'un ion positif, soit inférieure à la somme de l'énergie libérée lors de la formation des ions négatifs et de l'énergie de leurs échanges mutuels. attirance.
Il est bien évident que la formation d'un ion positif à partir d'un atome neutre nécessite le moins de travail dans le cas où se produisent des électrons situés dans la couche électronique qui a commencé à se former.
D’un autre côté, la plus grande énergie est libérée lorsqu’un électron s’attache à des atomes d’halogène, auxquels il manque un électron avant de remplir la couche électronique. Par conséquent, une liaison ionique se forme par transfert d’électrons, ce qui conduit à la création de couches électroniques remplies dans les ions résultants.
Un autre type de connexion - une liaison covalente.
Lorsque des molécules constituées d’atomes identiques se forment, la formation d’ions de charges opposées est impossible. La liaison ionique n’est donc pas possible. Cependant, dans la nature, il existe des substances dont les molécules sont formées d'atomes identiques. H2, O2, N2 etc. La liaison dans des substances de ce type est appelée covalent ou homéopolaire(homéo – différent [grec]). De plus, des liaisons covalentes sont également observées dans des molécules comportant des atomes différents : fluorure d'hydrogène HF, l'oxyde nitrique NON, méthane CH4 etc.
La nature des liaisons covalentes ne peut être expliquée que sur la base de la mécanique quantique. L’explication de la mécanique quantique repose sur la nature ondulatoire de l’électron. La fonction d'onde des électrons externes d'un atome ne s'arrête pas brusquement à mesure que la distance par rapport au centre de l'atome augmente, mais diminue progressivement. À mesure que les atomes se rapprochent, les nuages d’électrons flous des électrons externes se chevauchent partiellement, provoquant leur déformation. Un calcul précis du changement d'état des électrons nécessite de résoudre l'équation des ondes de Schrödinger pour le système de toutes les particules participant à l'interaction. La complexité et la lourdeur de cette voie nous obligent ici à nous limiter à une seule considération qualitative des phénomènes.
 Dans le cas le plus simple s-état d’un électron, le nuage d’électrons est une sphère d’un certain rayon. Si les deux électrons d'une molécule covalente échangent leurs places pour que l'électron 1, appartenant auparavant au noyau " UN", se déplacera à la place de l'électron 2, qui appartenait au noyau " b", et l'électron 2 fait une transition inverse, alors rien ne changera dans l'état de la molécule covalente.
Dans le cas le plus simple s-état d’un électron, le nuage d’électrons est une sphère d’un certain rayon. Si les deux électrons d'une molécule covalente échangent leurs places pour que l'électron 1, appartenant auparavant au noyau " UN", se déplacera à la place de l'électron 2, qui appartenait au noyau " b", et l'électron 2 fait une transition inverse, alors rien ne changera dans l'état de la molécule covalente.
Le principe de Pauli permet l’existence de deux électrons dans le même état et de spins opposés. La fusion des régions où les deux électrons peuvent être localisés signifie l'émergence entre eux d'une mécanique quantique particulière. interaction d'échange. Dans ce cas, chacun des électrons de la molécule peut appartenir alternativement à l'un ou l'autre noyau.
Comme le montrent les calculs, l'énergie d'échange d'une molécule est positive si les spins des électrons en interaction sont parallèles, et négative s'ils ne le sont pas.
Ainsi, la liaison de type covalente est assurée par une paire d’électrons de spins opposés. Si dans une liaison ionique on parlait du transfert d'électrons d'un atome à un autre, alors ici la connexion s'effectue en généralisant les électrons et en créant un espace commun pour leur mouvement.
Spectres moléculaires
Les spectres moléculaires sont très différents des spectres atomiques. Alors que les spectres atomiques sont constitués de raies individuelles, les spectres moléculaires sont constitués de bandes nettes à une extrémité et floues à l’autre. Par conséquent, les spectres moléculaires sont également appelés spectres rayés.
Des bandes dans les spectres moléculaires sont observées dans les gammes de fréquences infrarouges, visibles et ultraviolettes des ondes électromagnétiques. Dans ce cas, les rayures sont disposées dans un certain ordre, formant une série de rayures. Il existe un certain nombre de séries dans le spectre.
La mécanique quantique fournit une explication de la nature des spectres moléculaires. L'interprétation théorique des spectres des molécules polyatomiques est très complexe. Nous nous limiterons à considérer uniquement les molécules diatomiques.
Plus tôt, nous avons noté que l'énergie d'une molécule dépend de la nature du mouvement des noyaux atomiques et identifié trois types de cette énergie : translationnelle, rotationnelle et vibratoire. De plus, l’énergie d’une molécule est également déterminée par la nature du mouvement des électrons. Ce type d'énergie est appelé énergie électronique et est une composante de l’énergie totale de la molécule.
L’énergie totale de la molécule est donc :
Un changement d'énergie de translation ne peut pas conduire à l'apparition d'une raie spectrale dans le spectre moléculaire, nous exclurons donc ce type d'énergie lors d'un examen plus approfondi des spectres moléculaires. Alors
D'après la règle de fréquence de Bohr ( III– Postulat de Bohr) la fréquence du quantum émis par une molécule lorsque son état énergétique change est égale à
![]() .
.
L'expérience et les études théoriques ont montré que
Par conséquent, avec des excitations faibles, cela change seulement, avec des excitations plus fortes – et avec des excitations encore plus fortes –. Discutons plus en détail des différents types de spectres moléculaires.
Spectre de rotation des molécules
Commençons par explorer l'absorption des ondes électromagnétiques avec de petites portions d'énergie. Jusqu'à ce que la valeur du quantum d'énergie devienne égale à la distance entre les deux niveaux les plus proches, la molécule n'absorbera pas. En augmentant progressivement la fréquence, on atteindra des quanta capables de faire passer une molécule d'une étape de rotation à une autre. Cela se produit dans la région des ondes infrarouges de l'ordre de 0,1 à 1 mm.
![]() ,
,
où et sont les valeurs du nombre quantique de rotation aux -ième et -ième niveaux d'énergie.
Nombres quantiques rotationnels et peuvent avoir des valeurs, c'est-à-dire leurs modifications possibles sont limitées par la règle de sélection
L'absorption d'un quantum par une molécule le transfère d'un niveau d'énergie de rotation à un autre, plus élevé, et conduit à l'apparition d'une raie spectrale dans le spectre d'absorption rotationnelle. À mesure que la longueur d’onde diminue (c’est-à-dire que le nombre change), de plus en plus de nouvelles raies du spectre d’absorption apparaissent dans cette région. La totalité de toutes les lignes donne une idée de la répartition des états d'énergie de rotation de la molécule.
Nous avons jusqu'à présent considéré le spectre d'absorption de la molécule. Le spectre d'émission de la molécule est également possible. L'apparition de raies dans le spectre d'émission rotationnelle est associée à la transition de la molécule du niveau d'énergie de rotation supérieur au niveau inférieur.
Les spectres de rotation permettent de déterminer avec une grande précision les distances interatomiques dans des molécules simples. Connaissant le moment d’inertie et la masse des atomes, il est possible de déterminer les distances entre atomes. Pour une molécule diatomique
Spectre vibration-rotation des molécules
L'absorption d'ondes électromagnétiques par une substance dans la région infrarouge d'une longueur d'onde de l'ordre du micron provoque des transitions entre les niveaux d'énergie vibratoire et conduit à l'apparition d'un spectre vibratoire de la molécule. Cependant, lorsque les niveaux d’énergie vibrationnelle d’une molécule changent, ses états d’énergie de rotation changent simultanément. Les transitions entre deux niveaux d'énergie vibratoire s'accompagnent de changements dans les états d'énergie de rotation. Dans ce cas, un spectre vibrationnel-rotationnel de la molécule apparaît.
Si une molécule vibre et tourne simultanément, alors son énergie sera déterminée par deux nombres quantiques et :
![]() .
.
En tenant compte des règles de sélection pour les deux nombres quantiques, nous obtenons la formule suivante pour les fréquences du spectre vibration-rotationnel (la formule précédente /h et rejetons le niveau d'énergie précédent, c'est-à-dire les termes entre parenthèses) :
![]() .
.
Dans ce cas, le signe (+) correspond aux transitions d'un niveau de rotation inférieur à un niveau de rotation supérieur, et le signe (-) correspond à la position opposée. La partie vibrationnelle de la fréquence détermine la région spectrale dans laquelle se situe la bande ; la partie rotative détermine la structure fine de la bande, c'est-à-dire division de raies spectrales individuelles.
Selon les concepts classiques, la rotation ou la vibration d'une molécule diatomique ne peut conduire à l'émission d'ondes électromagnétiques que si la molécule a un moment dipolaire non nul. Cette condition n'est satisfaite que pour les molécules formées de deux atomes différents, c'est-à-dire pour les molécules asymétriques.
Une molécule symétrique formée d’atomes identiques a un moment dipolaire nul. Par conséquent, selon l’électrodynamique classique, la vibration et la rotation d’une telle molécule ne peuvent pas provoquer de rayonnement. La théorie quantique conduit à des résultats similaires.
Spectre vibrationnel électronique des molécules
L'absorption des ondes électromagnétiques dans le domaine visible et ultraviolet conduit à des transitions de la molécule entre différents niveaux d'énergie électronique, c'est-à-dire à l'apparition du spectre électronique de la molécule. Chaque niveau d'énergie électronique correspond à une certaine répartition spatiale des électrons ou, comme on dit, à une certaine configuration d'électrons à énergie discrète. Chaque configuration d'électrons correspond à de nombreux niveaux d'énergie vibratoire.
Une transition entre deux niveaux électroniques s’accompagne de nombreuses transitions entre niveaux vibratoires. C'est ainsi qu'apparaît le spectre vibratoire électronique de la molécule, constitué de groupes de raies proches.
Un système de niveaux de rotation est superposé à chaque état d’énergie vibratoire. Par conséquent, la fréquence d’un photon lors d’une transition électronique-vibratoire sera déterminée par un changement dans les trois types d’énergie :
![]() .
.
Fréquence - détermine la position du spectre.
L'ensemble du spectre vibratoire électronique est un système de plusieurs groupes de bandes, se chevauchant souvent et constituant une large bande.
L'étude et l'interprétation des spectres moléculaires permettent de comprendre la structure détaillée des molécules et sont largement utilisées pour l'analyse chimique.
diffusion Raman
Ce phénomène réside dans le fait que dans le spectre de diffusion qui se produit lorsque la lumière traverse des gaz, des liquides ou des corps cristallins transparents, ainsi que la diffusion de la lumière à fréquence constante, un certain nombre de fréquences supérieures ou inférieures apparaissent, correspondant aux fréquences de transitions vibrationnelles ou rotationnelles des molécules diffusantes.
Le phénomène de diffusion Raman a une explication simple en mécanique quantique. Le processus de diffusion de la lumière par les molécules peut être considéré comme une collision inélastique de photons avec des molécules. Lors d'une collision, un photon ne peut donner ou recevoir d'une molécule que des quantités d'énergie égales aux différences entre ses deux niveaux d'énergie. Si, lors d'une collision avec un photon, une molécule passe d'un état d'énergie inférieure à un état d'énergie plus élevée, elle perd son énergie et sa fréquence diminue. Cela crée une raie dans le spectre de la molécule, décalée par rapport à la raie principale vers des longueurs d'onde plus longues. Si, après une collision avec un photon, une molécule passe d'un état d'énergie plus élevée à un état d'énergie plus faible, une raie se crée dans le spectre qui est décalée par rapport à la principale vers des longueurs d'onde plus courtes.
Les études de diffusion Raman fournissent des informations sur la structure des molécules. Grâce à cette méthode, les fréquences de vibration naturelles des molécules sont déterminées facilement et rapidement. Cela permet également de juger de la nature de la symétrie de la molécule.
Luminescence
Si les molécules d'une substance peuvent être amenées dans un état excité sans augmenter leur énergie cinétique moyenne, c'est à dire sans chauffage, alors la lueur de ces corps ou luminescence se produit.
Il existe deux types de luminescence : fluorescence Et phosphorescence.
Fluorescence appelée luminescence, qui s'arrête immédiatement après la fin de l'action de l'excitateur de luminescence.
Avec la fluorescence, une transition spontanée des molécules d'un état excité vers un niveau inférieur se produit. Ce type de lueur a une durée très courte (environ 10 à 7 secondes).
Phosphorescence appelé luminescence, qui conserve son éclat longtemps après l'action de l'excitateur de luminescence.
Lors de la phosphorescence, une molécule passe d’un état excité à un niveau métastable. Métastable Il s’agit d’un niveau à partir duquel une transition vers un niveau inférieur est peu probable. Une émission peut se produire si la molécule revient à nouveau au niveau excité.
Le passage d'un état métastable à un état excité n'est possible qu'en présence d'une excitation supplémentaire. Un tel agent pathogène supplémentaire peut être la température de la substance. À haute température, cette transition se produit rapidement, à basse température, elle se produit lentement.
Comme nous l'avons déjà noté, la luminescence sous l'influence de la lumière est appelée photoluminescence, sous l’influence d’un bombardement électronique – cathodoluminescence, sous l’influence d’un champ électrique – électroluminescence, sous l'influence de transformations chimiques - chimiluminescence.
Amplificateurs quantiques et générateurs de rayonnement
Au milieu des années 50 de notre siècle, le développement rapide de l'électronique quantique a commencé. En 1954, les travaux des académiciens N.G. Basov et A.M. paraissent en URSS. Prokhorov, dans lequel un générateur quantique d'ondes radio ultracourtes de l'ordre du centimètre, appelé maître(amplification par micrologiciel par émission stimulée de rayonnement). Une série de générateurs et d'amplificateurs de lumière dans les domaines visible et infrarouge, apparus dans les années 60, furent appelés générateurs quantiques optiques ou lasers(Amplification de la lumière par émission stimulée de rayonnement).
Les deux types d'appareils fonctionnent sur la base de l'effet d'un rayonnement stimulé ou stimulé.
Examinons ce type de rayonnement plus en détail.
Ce type de rayonnement est le résultat de l’interaction d’une onde électromagnétique avec les atomes de la substance traversée par l’onde.
Dans les atomes, les transitions des niveaux d'énergie supérieurs aux niveaux inférieurs se produisent spontanément (ou spontanément). Cependant, sous l’influence du rayonnement incident, de telles transitions sont possibles à la fois dans le sens direct et dans le sens inverse. Ces transitions sont appelées forcé ou induit. Lors d'une transition forcée d'un des niveaux excités à un niveau d'énergie faible, l'atome émet un photon qui s'ajoute au photon sous l'influence duquel la transition s'est effectuée.
Dans ce cas, la direction de propagation de ce photon et, par conséquent, de tout rayonnement stimulé coïncide avec la direction de propagation du rayonnement externe qui a provoqué la transition, c'est-à-dire l'émission stimulée est strictement cohérente avec l'émission motrice.
Ainsi, le nouveau photon issu de l'émission stimulée amplifie la lumière traversant le milieu. Cependant, simultanément à l'émission induite, le processus d'absorption de la lumière se produit, car Le photon moteur est absorbé par un atome situé à un niveau d’énergie faible et l’atome se déplace vers un niveau d’énergie plus élevé. Et
Le processus de transfert de l'environnement vers un état inverse est appelé pompé environnement améliorant. Il existe de nombreuses méthodes pour pomper un milieu à gain. Le plus simple d'entre eux est le pompage optique d'un milieu, dans lequel les atomes sont transférés d'un niveau inférieur à un niveau excité supérieur en irradiant une lumière d'une fréquence telle que .
Dans un milieu à état inverse, l'émission stimulée dépasse l'absorption de la lumière par les atomes, ce qui entraîne une amplification du faisceau lumineux incident.
Considérons un dispositif utilisant de tels supports, utilisé comme générateur d'ondes dans le domaine optique ou laser.
 Sa partie principale est un cristal de rubis artificiel, qui est un oxyde d'aluminium dans lequel certains atomes d'aluminium sont remplacés par des atomes de chrome. Lorsqu'un cristal de rubis est irradié avec une lumière d'une longueur d'onde de 5 600, les ions chrome se déplacent vers le niveau d'énergie supérieur.
Sa partie principale est un cristal de rubis artificiel, qui est un oxyde d'aluminium dans lequel certains atomes d'aluminium sont remplacés par des atomes de chrome. Lorsqu'un cristal de rubis est irradié avec une lumière d'une longueur d'onde de 5 600, les ions chrome se déplacent vers le niveau d'énergie supérieur.
La transition de retour à l’état fondamental se déroule en deux étapes. Dans un premier temps, les ions excités cèdent une partie de leur énergie au réseau cristallin et entrent dans un état métastable. Les ions restent à ce niveau plus longtemps qu’au niveau supérieur. En conséquence, un état inverse d’un niveau métastable est obtenu.
 |
Le retour des ions à l'état fondamental s'accompagne de l'émission de deux raies rouges : et . Ce retour se produit comme une avalanche sous l'influence de photons de même longueur d'onde, c'est-à-dire à émission stimulée. Ce retour se produit beaucoup plus rapidement qu'avec une émission spontanée, la lumière est donc amplifiée.
Le rubis utilisé dans le laser a la forme d'une tige d'un diamètre de 0,5 cm et d'une longueur de 4 à 5 cm. Les extrémités plates de cette tige sont polies et argentées de manière à former deux miroirs se faisant face, l'un d'entre eux étant translucides. L'ensemble de la tige de rubis est situé à proximité d'un tube électronique pulsé, qui sert à pomper optiquement le milieu. Les photons dont les directions de mouvement forment de petits angles avec l'axe du rubis subissent de multiples réflexions depuis ses extrémités.
Par conséquent, leur chemin dans le cristal sera très long et les cascades de photons dans cette direction recevront le plus grand développement.
Les photons émis spontanément dans d'autres directions sortent du cristal par sa surface latérale sans provoquer de rayonnement supplémentaire.
Lorsque le faisceau axial devient suffisamment intense, une partie sort par l’extrémité translucide du cristal vers l’extérieur.
Une grande quantité de chaleur est générée à l’intérieur du cristal. Il doit donc être intensément refroidi.
Le rayonnement laser présente un certain nombre de caractéristiques. Il se caractérise par :
1. cohérence temporelle et spatiale ;
2. monochromatique strict ;
3. puissance élevée ;
4. étroitesse du faisceau.
La haute cohérence du rayonnement ouvre de larges perspectives pour l'utilisation des lasers pour les communications radio, en particulier pour les communications radio directionnelles dans l'espace. Si l’on trouve un moyen de moduler et de démoduler la lumière, il sera possible de transmettre une énorme quantité d’informations. Ainsi, en termes de volume d'informations transmises, un seul laser pourrait remplacer l'ensemble du système de communication entre les côtes est et ouest des États-Unis.
La largeur angulaire du faisceau laser est si petite qu'en utilisant une focalisation télescopique, il est possible d'obtenir un point lumineux d'un diamètre de 3 km sur la surface lunaire. La puissance élevée et l'étroitesse du faisceau permettent, lors de la focalisation à l'aide d'une lentille, d'obtenir une densité de flux énergétique 1000 fois supérieure à la densité de flux énergétique pouvant être obtenue en focalisant la lumière solaire. De tels faisceaux lumineux peuvent être utilisés pour l’usinage et le soudage, pour influencer le déroulement de réactions chimiques, etc.
Ce qui précède n’épuise pas toutes les capacités du laser. Il s'agit d'un type de source lumineuse complètement nouveau et il est encore difficile d'imaginer tous les domaines possibles de son application.
SPECTRES MOLÉCULAIRES, spectres d’émission et d’absorption électromagnétiques. rayonnement et combinaison diffusion de la lumière appartenant à des molécules libres ou faiblement liées. Ils ressemblent à un ensemble de bandes (lignes) dans les régions du spectre des rayons X, UV, visibles, IR et radio (y compris les micro-ondes). La position des bandes (lignes) dans les spectres d'émission (spectres moléculaires d'émission) et d'absorption (spectres moléculaires d'absorption) est caractérisée par des fréquences v (longueurs d'onde l = c/v, où c est la vitesse de la lumière) et des nombres d'ondes = 1. /l; il est déterminé par la différence entre les énergies E" et E : les états de la molécule entre lesquels se produit une transition quantique :
![]()
(h-constante de Planck). Avec combinaison En diffusion, la valeur hv est égale à la différence des énergies des photons incidents et diffusés. L'intensité des bandes (lignes) est liée au nombre (concentration) de molécules d'un type donné, à la population de niveaux d'énergie E" et E : et à la probabilité de la transition correspondante.
La probabilité de transitions avec l'émission ou l'absorption de rayonnement est déterminée principalement par le carré de l'élément électrique de la matrice. moment dipolaire de transition, et avec une considération plus précise - par les carrés des éléments de la matrice magnétique. et électrique moments quadripolaires de la molécule (voir Transitions quantiques). Avec combinaison En diffusion de la lumière, la probabilité de transition est liée à l'élément de matrice du moment dipolaire de transition induit de la molécule, c'est-à-dire avec l'élément matriciel de la polarisabilité de la molécule.
Les conditions disent. les systèmes, dont les transitions apparaissent sous la forme de certains spectres moléculaires, ont une nature différente et diffèrent grandement en énergie. Les niveaux d'énergie de certains types sont éloignés les uns des autres, de sorte que lors des transitions, la molécule absorbe ou émet un rayonnement haute fréquence. La distance entre les niveaux d'une autre nature est faible, et dans certains cas, en l'absence d'éléments externes. les niveaux de champ fusionnent (dégénérent). Avec de petites différences d'énergie, des transitions sont observées dans la région des basses fréquences. Par exemple, les noyaux des atomes de certains éléments ont le leur. mag. couple et électrique moment quadripolaire associé au spin. Les électrons ont aussi un champ magnétique moment associé à leur rotation. En l'absence d'intervention externe champs d'orientation magnétique les moments sont arbitraires, c'est-à-dire ils ne sont pas quantifiés et les énergies correspondantes. les États sont dégénérés. Lors de l'application externe aimant permanent champ, la dégénérescence est levée et des transitions entre les niveaux d’énergie sont possibles, observées dans la région des radiofréquences du spectre. C'est ainsi qu'apparaissent les spectres RMN et RPE (voir Résonance magnétique nucléaire, Résonance paramagnétique électronique).
Distribution cinétique énergies des électrons émis par mol. systèmes à la suite d'une irradiation avec des rayons X ou un rayonnement UV dur, donne des rayons Xspectroscopie et spectroscopie photoélectronique. Supplémentaire processus dans la jetée Le système, provoqué par l’excitation initiale, conduit à l’apparition d’autres spectres. Ainsi, les spectres Auger résultent de la relaxation. capture d'électrons externes coquilles de k.-l. atome par poste interne vacant coquille, et l’énergie libérée se transforme. en cinétique énergie d'un autre électron ext. coquille émise par un atome. Dans ce cas, une transition quantique se produit d'un certain état d'une molécule neutre à un état de mole. ion (voir spectroscopie Auger).
Traditionnellement, seuls les spectres associés aux spectres optiques sont classés comme spectres moléculaires proprement dits. transitions entre électronique-vibration-rotation, niveaux d'énergie d'une molécule associée à trois bases. types d'énergie niveaux de la molécule - électronique E el, vibrationnel E compte et rotationnel E bp, correspondant à trois types d'internes. mouvement dans une molécule. L'énergie de la configuration d'équilibre d'une molécule dans un état électronique donné est prise comme Anguille. L'ensemble des états électroniques possibles d'une molécule est déterminé par les propriétés de sa coque électronique et sa symétrie. Oscillation les mouvements des noyaux d'une molécule par rapport à leur position d'équilibre dans chaque état électronique sont quantifiés de telle sorte que pour plusieurs vibrations. degrés de liberté, un système complexe d'oscillations se forme. les niveaux d'énergie E comptent. La rotation de la molécule dans son ensemble en tant que système rigide de noyaux connectés est caractérisée par la rotation. moment de la quantité de mouvement, qui est quantifié, formant une rotation. états (niveaux d’énergie de rotation) E temps. Généralement, l’énergie des transitions électroniques est de l’ordre de plusieurs. eV, vibrationnel - 10 -2 ... 10 -1 eV, rotationnel - 10 -5 ... 10 -3 eV.
Selon les niveaux d'énergie, des transitions se produisent avec émission, absorption ou combinaisons. diffusion électromagnétique rayonnement - électronique, oscillation. ou rotationnelles, il y a des oscillations électroniques. et spectres moléculaires de rotation. Les articles Spectres électroniques, Spectres vibrationnels, Spectres rotationnels fournissent des informations sur les états correspondants des molécules, les règles de sélection des transitions quantiques, mol. spectroscopie, ainsi que les caractéristiques des molécules qui peuvent être utilisées. obtenus à partir de spectres moléculaires : propriétés et symétrie des états électroniques, vibrations. constantes, énergie de dissociation, symétrie de la molécule, rotation. constantes, moments d'inertie, géom. paramètres électriques moments dipolaires, données structurelles et internes champs de force, etc. Les spectres d'absorption électronique et de luminescence dans les régions visible et UV fournissent des informations sur la distribution
Spectre est une séquence de quanta d'énergie de rayonnement électromagnétique absorbé, libéré, diffusé ou réfléchi par une substance lors des transitions d'atomes et de molécules d'un état énergétique à un autre.
Selon la nature de l'interaction de la lumière avec la matière, les spectres peuvent être divisés en spectres d'absorption ; émissions (émission); diffusion et réflexion.
Pour les objets étudiés, la spectroscopie optique, c'est-à-dire spectroscopie dans la gamme de longueurs d'onde 10 -3 ÷10 -8 m divisé en atomique et moléculaire.
Spectre atomique est une séquence de lignes dont la position est déterminée par l'énergie de transition électronique d'un niveau à un autre.
Énergie atomique peut être représenté comme la somme de l'énergie cinétique du mouvement de translation et de l'énergie électronique :
où est la fréquence, est la longueur d’onde, est le nombre d’onde, est la vitesse de la lumière, est la constante de Planck.
Puisque l’énergie d’un électron dans un atome est inversement proportionnelle au carré du nombre quantique principal, l’équation d’une raie dans le spectre atomique peut s’écrire :
 . .
| (4.12) |
Ici  - les énergies des électrons à des niveaux supérieurs et inférieurs ; - constante de Rydberg ;
- les énergies des électrons à des niveaux supérieurs et inférieurs ; - constante de Rydberg ;  - termes spectraux exprimés en unités de nombres d'ondes (m -1, cm -1).
- termes spectraux exprimés en unités de nombres d'ondes (m -1, cm -1).
Toutes les raies du spectre atomique convergent dans la région des ondes courtes jusqu'à une limite déterminée par l'énergie d'ionisation de l'atome, après quoi il existe un spectre continu.
Énergie moléculaire en première approximation, il peut être considéré comme la somme des énergies de translation, de rotation, de vibration et électroniques :
 | (4.15) |
Pour la plupart des molécules, cette condition est satisfaite. Par exemple, pour H 2 à 291 K, les composantes individuelles de l'énergie totale diffèrent d'un ordre de grandeur ou plus :
309,5 kJ/mole,
 =25,9 kJ/mole,
=25,9 kJ/mole,
2,5 kJ/mole,
 =3,8 kJ/mol.
=3,8 kJ/mol.
Les valeurs énergétiques des quanta dans différentes régions du spectre sont comparées dans le tableau 4.2.
Tableau 4.2 - Énergie des quanta absorbés de différentes régions du spectre optique des molécules
Les notions de « vibrations des noyaux » et de « rotation des molécules » sont relatives. En réalité, de tels types de mouvements ne véhiculent que de manière très approximative des idées sur la répartition des noyaux dans l’espace, qui est de même nature probabiliste que la répartition des électrons.
Un système schématique de niveaux d'énergie dans le cas d'une molécule diatomique est présenté à la figure 4.1.
Les transitions entre les niveaux d'énergie de rotation conduisent à l'apparition de spectres de rotation dans les régions de l'IR lointain et des micro-ondes. Les transitions entre les niveaux vibrationnels au sein d'un même niveau électronique donnent des spectres vibration-rotation dans la région proche infrarouge, puisqu'un changement dans le nombre quantique vibrationnel entraîne inévitablement un changement dans le nombre quantique rotation. Enfin, les transitions entre niveaux électroniques provoquent l’apparition de spectres électronique-vibration-rotation dans les domaines visible et UV.
Dans le cas général, le nombre de transitions peut être très important, mais en fait toutes n’apparaissent pas dans les spectres. Le nombre de transitions est limité règles de sélection .
Les spectres moléculaires fournissent une mine d’informations. Ils peuvent être utilisés :
Identifier les substances dans l'analyse qualitative, car chaque substance a son propre spectre unique ;
Pour l'analyse quantitative ;
Pour l'analyse de groupes structuraux, puisque certains groupes, tels que >C=O, _ NH 2, _ OH, etc., donnent des bandes caractéristiques dans les spectres ;
Déterminer les états énergétiques des molécules et les caractéristiques moléculaires (distance internucléaire, moment d'inertie, fréquences de vibration naturelles, énergies de dissociation) ; une étude approfondie des spectres moléculaires permet de tirer des conclusions sur la structure spatiale des molécules ;
Dans les études cinétiques, notamment pour l'étude de réactions très rapides.

 - l'énergie des niveaux électroniques ;
- l'énergie des niveaux électroniques ;
Énergie des niveaux vibratoires ;
Énergies des niveaux de rotation
Figure 4.1 – Disposition schématique des niveaux d'énergie d'une molécule diatomique
Loi Bouguer-Lambert-Bière
La base de l'analyse moléculaire quantitative utilisant la spectroscopie moléculaire est Loi Bouguer-Lambert-Bière , reliant l'intensité de la lumière incidente et transmise à la concentration et à l'épaisseur de la couche absorbante (Figure 4.2) :
ou avec un facteur de proportionnalité :
Résultat de l'intégration :
 | (4.19) |
 . .
| (4.20) |
Lorsque l'intensité de la lumière incidente diminue d'un ordre de grandeur
 . .
| (4.21) |
Si =1 mol/l, alors, soit Le coefficient d'absorption est égal à l'épaisseur réciproque de la couche dans laquelle, à une concentration égale à 1, l'intensité de la lumière incidente diminue d'un ordre de grandeur.
Coefficients d'absorption et dépendent de la longueur d'onde. Le type de cette dépendance est une sorte d'« empreinte digitale » de molécules, qui est utilisée dans l'analyse qualitative pour identifier une substance. Cette dépendance est caractéristique et individuelle pour une substance particulière et reflète les groupes et liaisons caractéristiques inclus dans la molécule.
Densité optique D
exprimé en %
4.2.3 Énergie de rotation d'une molécule diatomique dans l'approximation du rotateur rigide. Spectres de rotation des molécules et leur application pour déterminer les caractéristiques moléculaires
L'apparition de spectres de rotation est due au fait que l'énergie de rotation de la molécule est quantifiée, c'est-à-dire
| 0 |
| UN |
Depuis le point Ô est le centre de gravité de la molécule, alors :
Introduction de la notation de masse réduite :
 | (4.34) |
conduit à l'équation
 . .
| (4.35) |
Ainsi, une molécule diatomique (Figure 4.7 UN), tournant autour d'un axe ou passant par le centre de gravité, peut être simplifié pour être considéré comme une particule de masse , décrivant un cercle de rayon autour du point Ô(Figure 4.7 b).
La rotation d'une molécule autour d'un axe donne un moment d'inertie pratiquement égal à zéro, puisque les rayons des atomes sont bien inférieurs à la distance internucléaire. La rotation autour des axes ou, mutuellement perpendiculaires à la ligne de liaison de la molécule, conduit à des moments d'inertie d'égale ampleur :
où est un nombre quantique rotationnel qui ne prend que des valeurs entières
0, 1, 2…. Conformément à règle de sélection pour le spectre de rotation d'une molécule diatomique, une modification du nombre quantique de rotation lors de l'absorption d'un quantum d'énergie n'est possible que par un, c'est-à-dire
transforme l'équation (4.37) sous la forme :
 20
20  12
6
2
12
6
2 |
numéro d'onde de la raie dans le spectre de rotation correspondant à l'absorption d'un quantum lors de la transition de j niveau d'énergie par niveau j+1, peut être calculé à l'aide de l'équation :
Ainsi, le spectre de rotation dans l’approximation du modèle de rotateur rigide est un système de lignes situées à la même distance les unes des autres (Figure 4.5b). Des exemples de spectres de rotation de molécules diatomiques estimés dans le modèle de rotateur rigide sont présentés dans la figure 4.6.
| UN b |


Figure 4.6 – Spectres de rotation HF (UN) Et CO(b)
Pour les molécules d'halogénure d'hydrogène, ce spectre est décalé vers la région IR lointaine du spectre, pour les molécules plus lourdes - vers le micro-ondes.
Sur la base des modèles obtenus d'apparition du spectre de rotation d'une molécule diatomique, en pratique, la distance entre les raies adjacentes du spectre est d'abord déterminée, à partir de laquelle elles sont ensuite trouvées, et à l'aide des équations :
 , ,
| (4.45) |
Où - constante de distorsion centrifuge
, est lié à la constante de rotation par la relation approximative  . La correction ne doit être prise en compte que pour les très grandes j.
. La correction ne doit être prise en compte que pour les très grandes j.
Pour les molécules polyatomiques, en général, trois moments d'inertie différents sont possibles  . S'il y a des éléments de symétrie dans la molécule, les moments d'inertie peuvent coïncider voire être égaux à zéro. Par exemple, pour les molécules polyatomiques linéaires(CO 2 , OCS, HCN, etc.)
. S'il y a des éléments de symétrie dans la molécule, les moments d'inertie peuvent coïncider voire être égaux à zéro. Par exemple, pour les molécules polyatomiques linéaires(CO 2 , OCS, HCN, etc.)
Où  - position de la ligne correspondant à la transition rotationnelle
- position de la ligne correspondant à la transition rotationnelle  dans une molécule isotopiquement substituée.
dans une molécule isotopiquement substituée.
Pour calculer l'ampleur du déplacement isotopique de la raie, il est nécessaire de calculer séquentiellement la masse réduite de la molécule isotopiquement substituée, en tenant compte du changement de la masse atomique de l'isotope, du moment d'inertie, de la constante de rotation et de la position. de la raie dans le spectre de la molécule selon les équations (4.34), (4.35), (4.39) et (4.43), respectivement, ou estimer le rapport des nombres d'onde des raies correspondant à la même transition en isotopiquement substitué et non -des molécules isotopiquement substituées, puis déterminer la direction et l'ampleur du déplacement isotopique à l'aide de l'équation (4.50). Si la distance internucléaire est approximativement considérée comme constante  , alors le rapport des nombres d'ondes correspond au rapport inverse des masses réduites :
, alors le rapport des nombres d'ondes correspond au rapport inverse des masses réduites :
où est le nombre total de particules, est le nombre de particules par je- ce niveau d'énergie à température T, k– constante de Boltzmann, - statistique forcer degré de dégénérescence je-de ce niveau d'énergie, caractérise la probabilité de trouver des particules à un niveau donné.
Pour un état de rotation, le niveau de population est généralement caractérisé par le rapport du nombre de particules j- ce niveau d'énergie au nombre de particules au niveau zéro :
 , ,
| (4.53) |
Où  - poids statistique j-de ce niveau d'énergie de rotation, correspond au nombre de projections de l'impulsion d'une molécule en rotation sur son axe - la ligne de communication de la molécule,
- poids statistique j-de ce niveau d'énergie de rotation, correspond au nombre de projections de l'impulsion d'une molécule en rotation sur son axe - la ligne de communication de la molécule,  , énergie de niveau de rotation nulle
, énergie de niveau de rotation nulle  . La fonction passe par un maximum à mesure qu'elle augmente j, comme illustré sur la figure 4.7 en utilisant la molécule de CO comme exemple.
. La fonction passe par un maximum à mesure qu'elle augmente j, comme illustré sur la figure 4.7 en utilisant la molécule de CO comme exemple.
L'extremum de la fonction correspond au niveau de population relative maximale dont la valeur du nombre quantique peut être calculée à l'aide de l'équation obtenue après détermination de la dérivée de la fonction à l'extremum :
 . .
| (4.54) |


Figure 4.7 – Population relative des niveaux d’énergie de rotation
molécules COà des températures de 298 et 1000 K
Exemple. Dans le spectre de rotation HI, la distance entre les lignes adjacentes est déterminée  cm -1. Calculez la constante de rotation, le moment d'inertie et la distance internucléaire d'équilibre dans la molécule.
cm -1. Calculez la constante de rotation, le moment d'inertie et la distance internucléaire d'équilibre dans la molécule.
Solution
Dans l'approximation du modèle de rotateur rigide, conformément à l'équation (4.45), nous déterminons la constante de rotation :
 cm -1.
cm -1.
Le moment d'inertie de la molécule est calculé à partir de la valeur de la constante de rotation à l'aide de l'équation (4.46) :
 kg . m2.
kg . m2.
Pour déterminer la distance internucléaire d'équilibre, nous utilisons l'équation (4.47), en tenant compte du fait que les masses des noyaux d'hydrogène  et de l'iode
et de l'iode  exprimé en kg :
exprimé en kg :
Exemple. Dans la région IR lointaine du spectre de 1 H 35 Cl, des raies ont été détectées dont les numéros d'onde sont :
Déterminer les valeurs moyennes du moment d'inertie et de la distance internucléaire de la molécule. Attribuez les raies observées dans le spectre aux transitions de rotation.
Solution
Selon le modèle du rotateur rigide, la différence des nombres d'ondes des raies adjacentes du spectre de rotation est constante et égale à 2. Déterminons la constante de rotation à partir de la valeur moyenne des distances entre raies adjacentes dans le spectre :
![]() cm-1,
cm-1,
 cm -1
cm -1
On retrouve le moment d'inertie de la molécule (équation (4.46)) :
Nous calculons la distance internucléaire d'équilibre (équation (4.47)), en tenant compte du fait que les masses des noyaux d'hydrogène  et du chlore
et du chlore  (exprimé en kg) :
(exprimé en kg) :
À l'aide de l'équation (4.43), nous estimons la position des raies dans le spectre de rotation de 1 H 35 Cl :
Comparons les valeurs calculées des nombres d'onde des lignes avec les valeurs expérimentales. Il s'avère que les raies observées dans le spectre de rotation de 1 H 35 Cl correspondent aux transitions :
| N lignes | |||||||
| , cm -1 | 85.384 | 106.730 | 128.076 | 149.422 | 170.768 | 192.114 | 213.466 |
 | 3 4 | 4 5 | 5 6 | 6 7 | 7 8 | 8 9 | 9 10 |
Exemple. Déterminer l'ampleur et la direction du déplacement isotopique de la raie d'absorption correspondant à la transition avec  niveau d'énergie dans le spectre de rotation de la molécule 1 H 35 Cl lorsque l'atome de chlore est remplacé par l'isotope 37 Cl. La distance internucléaire dans les molécules 1 H 35 Cl et 1 H 37 Cl est considérée comme la même.
niveau d'énergie dans le spectre de rotation de la molécule 1 H 35 Cl lorsque l'atome de chlore est remplacé par l'isotope 37 Cl. La distance internucléaire dans les molécules 1 H 35 Cl et 1 H 37 Cl est considérée comme la même.
Solution
Déterminer l'ampleur du déplacement isotopique de la raie correspondant à la transition , on calcule la masse réduite de la molécule 1 H 37 Cl en tenant compte de l'évolution de la masse atomique du 37 Cl :
Ensuite, nous calculons le moment d'inertie, la constante de rotation et la position de la ligne.  dans le spectre de la molécule 1 H 37 Cl et la valeur du déplacement isotopique selon les équations (4.35), (4.39), (4.43) et (4.50), respectivement.
dans le spectre de la molécule 1 H 37 Cl et la valeur du déplacement isotopique selon les équations (4.35), (4.39), (4.43) et (4.50), respectivement.
Autrement, le déplacement isotopique peut être estimé à partir du rapport des nombres d’onde des raies correspondant à la même transition dans les molécules (on suppose que la distance internucléaire est constante) puis de la position de la raie dans le spectre en utilisant l’équation (4.51).
Pour les molécules 1 H 35 Cl et 1 H 37 Cl, le rapport des nombres d'onde d'une transition donnée est égal à :

Pour déterminer le numéro d'onde de la raie d'une molécule isotopiquement substituée, on substitue la valeur du numéro d'onde de transition trouvé dans l'exemple précédent j → j+1 (3→4):
Nous concluons : le déplacement isotopique vers la région des basses fréquences ou des ondes longues est
85,384-83,049=2,335 cm-1.
Exemple. Calculez le nombre d'onde et la longueur d'onde de la raie spectrale la plus intense du spectre de rotation de la molécule 1 H 35 Cl. Faites correspondre la ligne avec la transition de rotation correspondante.
Solution
La raie la plus intense du spectre de rotation d’une molécule est associée à la population relative maximale du niveau d’énergie de rotation.
Substitution de la valeur de la constante de rotation trouvée dans l'exemple précédent pour 1 H 35 Cl (  cm -1) dans l'équation (4.54) permet de calculer le numéro de ce niveau d'énergie :
cm -1) dans l'équation (4.54) permet de calculer le numéro de ce niveau d'énergie :
 .
.
Le nombre d'onde de la transition rotationnelle à partir de ce niveau est calculé à l'aide de l'équation (4.43) :
On retrouve la longueur d'onde de transition de l'équation (4.11) transformée par rapport à :
4.2.4 Tâche multivariée n°11 « Spectres de rotation des molécules diatomiques »
1. Écrivez une équation de mécanique quantique pour calculer l’énergie du mouvement de rotation d’une molécule diatomique en tant que rotateur rigide.
2. Dérivez une équation pour calculer le changement d'énergie de rotation d'une molécule diatomique en tant que rotateur rigide lors de sa transition vers un niveau quantique adjacent plus élevé.  .
.
3. Dérivez une équation pour la dépendance du nombre d'onde des raies de rotation dans le spectre d'absorption d'une molécule diatomique sur le nombre quantique de rotation.
4. Dérivez une équation pour calculer la différence des nombres d’onde des raies voisines dans le spectre d’absorption rotationnelle d’une molécule diatomique.
5. Calculer la constante de rotation (en cm -1 et m -1) de la molécule diatomique UN par les numéros d'onde de deux raies adjacentes dans la région infrarouge à ondes longues du spectre d'absorption rotationnelle de la molécule (voir tableau 4.3).
6. Déterminer l'énergie de rotation de la molécule UN aux cinq premiers niveaux de rotation quantique (J).
7. Dessinez schématiquement les niveaux d’énergie du mouvement de rotation d’une molécule diatomique en tant que rotateur rigide.
8. Dessinez en pointillés sur ce diagramme les niveaux quantiques de rotation d’une molécule qui n’est pas un rotateur rigide.
9. Dérivez une équation pour calculer la distance internucléaire d'équilibre en fonction de la différence entre les nombres d'ondes des raies voisines dans le spectre d'absorption rotationnelle.
10. Déterminer le moment d'inertie (kg. m2) d'une molécule diatomique UN.
11. Calculer la masse réduite (kg) de la molécule UN.
12. Calculer la distance internucléaire d'équilibre () de la molécule UN. Comparez la valeur obtenue avec les données de référence.
13. Attribuer les raies observées dans le spectre de rotation de la molécule UN aux transitions de rotation.
14. Calculer le numéro d'onde de la raie spectrale correspondant à la transition rotationnelle du niveau j pour une molécule UN(voir tableau 4.3).
15. Calculer la masse réduite (kg) de la molécule isotopiquement substituée B.
16. Calculer le numéro d'onde de la raie spectrale associée à la transition rotationnelle à partir du niveau j pour une molécule B(voir tableau 4.3). Distances internucléaires dans les molécules UN Et B considérer comme égal.
17. Déterminer l'ampleur et la direction du déplacement isotopique dans les spectres de rotation des molécules UN Et B pour la raie spectrale correspondant à la transition de niveau rotationnelle j.
18. Expliquez la raison du changement non monotone de l'intensité des raies d'absorption à mesure que l'énergie de rotation de la molécule augmente
19. Déterminer le nombre quantique du niveau de rotation correspondant à la population relative la plus élevée. Calculer les longueurs d'onde des raies spectrales les plus intenses des spectres de rotation des molécules UN Et B.
Outre les spectres correspondant au rayonnement d'atomes individuels, des spectres émis par des molécules entières sont également observés (§ 61). Les spectres moléculaires ont une structure beaucoup plus diversifiée et complexe que les spectres atomiques. Ici, on observe des séquences condensées de raies, semblables aux séries spectrales des atomes, mais avec une loi de fréquence différente et avec des raies si rapprochées qu'elles se fondent en bandes continues (Fig. 279). En raison de la nature particulière de ces spectres, ils sont appelés rayés.
Riz. 279. Spectre rayé
Parallèlement à cela, des séquences de raies spectrales équidistantes et, enfin, des spectres multilignes sont observées, dans lesquelles, à première vue, il est difficile d'établir des modèles (Fig. 280). Il convient de noter que lors de l’étude du spectre de l’hydrogène, nous avons toujours une superposition du spectre moléculaire de Ha sur le spectre atomique et que des mesures spéciales doivent être prises pour augmenter l’intensité des raies émises par les atomes d’hydrogène individuels.

Riz. 280. Spectre moléculaire de l'hydrogène
D'un point de vue quantique, comme dans le cas des spectres atomiques, chaque raie du spectre moléculaire est émise lorsqu'une molécule passe d'un niveau d'énergie stationnaire à un autre. Mais dans le cas d’une molécule, il existe bien d’autres facteurs dont dépend l’énergie de l’état stationnaire.
Dans le cas le plus simple d'une molécule diatomique, l'énergie est composée de trois parties : 1) l'énergie de la couche électronique de la molécule ; 2) l'énergie des vibrations des noyaux des atomes qui composent la molécule le long de la ligne droite les reliant ; 3) l'énergie de rotation des noyaux autour d'un centre de masse commun. Les trois types d’énergie sont quantifiés, c’est-à-dire qu’ils ne peuvent prendre qu’une série discrète de valeurs. La couche électronique d'une molécule est formée à la suite de la fusion des couches électroniques des atomes qui composent la molécule. Les états électroniques énergétiques des molécules peuvent être considérés comme un cas limite
un effet Stark très fort provoqué par l'interaction interatomique des atomes formant une molécule. Bien que les forces qui lient les atomes en molécules soient de nature purement électrostatique, une compréhension correcte de la liaison chimique s'est avérée possible uniquement dans le cadre de la théorie quantique moderne de la mécanique ondulatoire.
Il existe deux types de molécules : homéopolaires et hétéropolaires. À mesure que la distance entre les noyaux augmente, les molécules homéopolaires se désintègrent en parties neutres. Les molécules hémopolaires comprennent des molécules. Les molécules hétéropolaires, à mesure que la distance entre les noyaux augmente, se désintègrent en ions positifs et négatifs. Un exemple typique de molécules hétéropolaires sont les molécules de sels, par exemple, etc. (vol. I, § 121, 130, 1959 ; dans l'édition précédente, § 115 et 124, etc. II, § 19, 22, 1959 ; dans l'édition précédente § 21 et 24).
Les états énergétiques du nuage électronique d’une molécule homéopolaire sont déterminés dans une large mesure par les propriétés ondulatoires des électrons.
Considérons un modèle très approximatif de la molécule la plus simple (une molécule d'hydrogène ionisée représentant deux « trous » potentiels situés à proximité l'un de l'autre et séparés par une « barrière » (Fig. 281).

Riz. 281. Deux trous potentiels.

Riz. 282. Fonctions d'onde d'un électron dans le cas de « puits » distants.
Chacun des « trous » représente l’un des atomes qui composent la molécule. Avec une grande distance entre les atomes, l'électron dans chacun d'eux a des valeurs d'énergie quantifiées correspondant aux ondes électroniques stationnaires dans chacun des « puits » séparément (§ 63). En figue. 282, a et b, deux fonctions d'onde identiques sont représentées qui décrivent l'état des électrons situés dans des atomes isolés. Ces fonctions d'onde correspondent au même niveau d'énergie.
Lorsque les atomes se rassemblent pour former une molécule, la « barrière » entre les « trous » devient « transparente » (§ 63), car sa largeur devient proportionnelle à la longueur de l’onde électronique. En conséquence, il y a
échange d'électrons entre atomes à travers une « barrière », et cela n'a aucun sens de parler de l'appartenance d'un électron à l'un ou l'autre atome.
La fonction d'onde peut désormais avoir deux formes : c et d (Fig. 283). Le cas c peut être approximativement considéré comme le résultat de l'addition des courbes a et b (Fig. 282), le cas comme la différence entre a et b, mais les énergies correspondant aux états c et d ne sont plus exactement égales entre elles. L'énergie de l'état est légèrement inférieure à l'énergie de l'état. Ainsi, de chaque niveau atomique naissent deux niveaux électroniques moléculaires.

Riz. 283. Fonctions d'onde d'un électron dans le cas de « puits » proches.
Jusqu’à présent, nous avons parlé de l’ion d’une molécule d’hydrogène, qui possède un électron. Une molécule d'hydrogène neutre possède deux électrons, ce qui conduit à devoir prendre en compte les positions relatives de leurs spins. Conformément au principe de Pauli, les électrons avec des spins parallèles semblent « s'éviter », donc la densité de probabilité de trouver chaque électron est distribuée selon la figure. 284, a, c'est-à-dire que les électrons sont le plus souvent situés en dehors de l'espace entre les noyaux. Par conséquent, avec des spins parallèles, une molécule stable ne peut pas être formée. Au contraire, les spins antiparallèles correspondent à la probabilité la plus élevée de trouver les deux électrons à l'intérieur de l'espace entre les noyaux (Fig. 294, b). Dans ce cas, la charge électronique négative attire à la fois les noyaux positifs et l’ensemble du système forme une molécule stable.
Dans les molécules hétéropolaires, le modèle de distribution de la densité de charge électronique est beaucoup plus classique. Un excès d'électrons se regroupe près de l'un des noyaux, tandis que près de l'autre, au contraire, il y a un manque d'électrons. Ainsi, deux ions se forment dans la molécule, positif et négatif, qui sont attirés l'un vers l'autre : par exemple, et
Le symbolisme des états électroniques des molécules présente de nombreuses similitudes avec le symbolisme atomique. Naturellement, dans une molécule, le rôle principal est joué par la direction de l'axe reliant les noyaux. Ici, le nombre quantique A est introduit, analogue à I dans l'atome. Le nombre quantique caractérise la valeur absolue de la projection sur l'axe de la molécule du moment orbital résultant du nuage électronique de la molécule.
Entre les valeurs et les symboles des états électroniques moléculaires, il existe une correspondance similaire à celle des atomes (§ 67) :

La valeur absolue de la projection du spin résultant du nuage électronique sur l'axe de la molécule est caractérisée par le nombre quantique 2, et la projection du moment de rotation total de la couche électronique est caractérisée par le nombre quantique.
Le nombre quantique est similaire au nombre quantique interne d'un atome (§59 et 67).

Riz. 284. Densité de probabilité de trouver un électron en différents points d'une molécule.
Tout comme les atomes, les molécules présentent une multiplicité causée par différentes orientations du spin résultant par rapport au moment orbital résultant.
Compte tenu de ces circonstances, les états électroniques des molécules s’écrivent comme suit :
où 5 est la valeur du spin résultant, et désigne l'un des symboles ou A, correspondant à différentes valeurs du nombre quantique A. Par exemple, l'état normal d'une molécule d'hydrogène est 2, l'état normal d'un hydroxyle La molécule est l'état normal d'une molécule d'oxygène. Lors des transitions entre différents états électroniques, les règles de sélection suivantes s'appliquent : .
L'énergie vibrationnelle d'une molécule associée aux vibrations des noyaux est quantifiée en tenant compte des propriétés ondulatoires des noyaux. En supposant que les noyaux d'une molécule sont liés par une force quasi-élastique (l'énergie potentielle d'une particule est proportionnelle au carré du déplacement, § 63), on obtient de l'équation de Schrödinger les valeurs autorisées suivantes de la vibration énergie de ce système (harmonique
oscillateur):
![]()
où est la fréquence des oscillations naturelles des noyaux, déterminée comme d'habitude (Vol. I, § 57, 1959 ; dans l'édition précédente § 67) :
![]()
où est la masse réduite des noyaux ; masses des deux noyaux ; constante quasi-élastique d'une molécule ; nombre quantique égal à En raison de la masse importante, la fréquence se situe dans la région infrarouge du spectre.

Riz. 285. Niveaux d'énergie vibratoire d'une molécule.
La constante quasi-élastique dépend de la configuration de la couche électronique et est donc différente pour les différents états électroniques de la molécule. Cette constante est d’autant plus grande que la molécule est forte, c’est-à-dire plus la liaison chimique est forte.
La formule (3) correspond à un système de niveaux d'énergie équidistants, dont la distance est de 1000. En fait, à de grandes amplitudes d'oscillations nucléaires, les écarts de la force de rappel par rapport à la loi de Hooke commencent déjà à se manifester. En conséquence, les niveaux d'énergie se rapprochent (Fig. 285). À des amplitudes suffisamment grandes, la molécule se dissocie en plusieurs parties.
Pour un oscillateur harmonique, les transitions ne sont autorisées qu'à , ce qui correspond à l'émission ou à l'absorption de lumière de fréquence. En raison des écarts par rapport à l'harmonicité, des transitions apparaissent qui correspondent à
Selon la condition quantique des fréquences (§ 58), des harmoniques devraient apparaître dans ce cas, ce qui est observé dans les spectres des molécules.
L'énergie vibratoire est un ajout relativement faible à l'énergie du nuage électronique d'une molécule. Les vibrations des noyaux conduisent au fait que chaque niveau électronique se transforme en un système de niveaux proches correspondant à différentes valeurs d'énergie vibratoire (Fig. 286). Cela n’épuise pas la complexité du système de niveaux d’énergie d’une molécule.

Riz. 286. Addition d'énergie vibratoire et électronique d'une molécule.
Il est également nécessaire de prendre en compte la plus petite composante de l'énergie moléculaire - l'énergie de rotation. Les valeurs admissibles de l'énergie de rotation sont déterminées, selon la mécanique des vagues, sur la base du principe de quantification du couple.
Selon la mécanique ondulatoire, le couple (§ 59) de tout système quantifié est égal à
![]()
Dans ce cas, remplace et est égal à 0, 1, 2, 3, etc.
Énergie cinétique d'un corps en rotation dans le précédent. éd. § 42) sera
![]()
où le moment d'inertie, co est la vitesse angulaire de rotation.
Mais par contre le couple est égal, on obtient donc :
ou, en substituant l'expression (5), on trouve finalement :
![]()
En figue. 287 montre les niveaux de rotation de la molécule ; contrairement aux niveaux vibrationnels et atomiques, la distance entre les niveaux de rotation augmente avec l'augmentation des transitions entre les niveaux de rotation sont autorisées et des raies avec des fréquences sont émises
![]()
où Evrash correspond correspond
La formule (9) donne les fréquences

Riz. 287. Niveaux d'énergie de rotation d'une molécule.
Nous obtenons des raies spectrales équidistantes situées dans la partie infrarouge lointain du spectre. La mesure des fréquences de ces raies permet de déterminer le moment d'inertie de la molécule. Il s'est avéré que les moments d'inertie des molécules sont de l'ordre de grandeur. Il est à noter que le moment d'inertie I lui-même dû au action
les forces centrifuges augmentent avec l'augmentation de la vitesse de rotation de la molécule. La présence de rotations conduit au fractionnement de chaque niveau d'énergie vibratoire en un certain nombre de sous-niveaux proches correspondant à différentes valeurs d'énergie de rotation.
Lorsqu'une molécule passe d'un état énergétique à un autre, les trois types d'énergie de la molécule peuvent changer simultanément (Fig. 288). En conséquence, chaque raie spectrale qui serait émise lors d’une transition électronique-vibratoire acquiert une fine structure rotationnelle et se transforme en une bande moléculaire typique.

Riz. 288. Changement simultané des trois types d'énergie d'une molécule
De telles bandes de lignes équidistantes sont observées dans la vapeur et l’eau et se situent dans la partie infrarouge lointain du spectre. On les observe non pas dans le spectre d'émission de ces vapeurs, mais dans leur spectre d'absorption, car les fréquences correspondant aux fréquences propres des molécules sont absorbées plus fortement que les autres. En figue. 289 montre une bande dans le spectre d'absorption de vapeur dans la région proche infrarouge. Cette bande correspond à des transitions entre des états énergétiques qui diffèrent non seulement en énergie de rotation, mais aussi en énergie vibrationnelle (à énergie constante des couches électroniques). Dans ce cas, et et Ecol changent simultanément, ce qui entraîne de grands changements d'énergie, c'est-à-dire que les raies spectrales ont une fréquence plus élevée que dans le premier cas considéré.
Conformément à cela, des raies apparaissent dans le spectre situé dans la région proche infrarouge, similaires à celles représentées sur la figure. 289.

Riz. 289. Bande d'absorption.
Le centre de la bande ( correspond à une transition à EUR constant ; selon la règle de sélection, de telles fréquences ne sont pas émises par la molécule. Les lignes avec des fréquences plus élevées - des longueurs d'onde plus courtes - correspondent à des transitions dans lesquelles le changement en EUR s'ajoute à Les lignes avec les fréquences les plus basses (côté droit) correspondent à la relation inverse : le changement d'énergie de rotation a le signe opposé.
A côté de ces bandes, on observe des bandes correspondant à des transitions avec un changement du moment d'inertie mais avec Dans ce cas, selon la formule (9), les fréquences des lignes doivent dépendre et les distances entre les lignes deviennent inégales. Chaque bande est constituée d'une série de lignes se condensant vers un bord,
qui s'appelle la tête de la bande. Pour la fréquence d'une raie spectrale individuelle incluse dans la bande, Delander a donné en 1885 une formule empirique de la forme suivante :
où est un entier.
La formule de Delandre découle directement des considérations ci-dessus. La formule de Delandre peut être représentée graphiquement si nous la traçons le long d'un axe et le long de l'autre (Fig. 290).

Riz. 290. Représentation graphique de la formule de Delandre.
Ci-dessous se trouvent les lignes correspondantes, formant, comme on le voit, une bande typique. La structure du spectre moléculaire dépendant fortement du moment d'inertie de la molécule, l'étude des spectres moléculaires est l'un des moyens fiables pour déterminer cette valeur. Les moindres changements dans la structure d’une molécule peuvent être détectés en étudiant son spectre. Le plus intéressant est le fait que les molécules contenant différents isotopes (§ 86) d'un même élément doivent avoir des raies différentes dans leur spectre, correspondant à différentes masses de ces isotopes. Cela découle du fait que les masses des atomes déterminent à la fois la fréquence de leurs vibrations dans la molécule et son moment d'inertie. En effet, les raies de bande du chlorure de cuivre sont constituées de quatre composants, correspondant à quatre combinaisons d'isotopes de cuivre 63 et 65 avec les isotopes de chlore 35 et 37 :
Des raies correspondant à des molécules contenant un isotope lourd de l'hydrogène ont également été découvertes, malgré le fait que la concentration de l'isotope dans l'hydrogène ordinaire est égale à
En plus de la masse des noyaux, d’autres propriétés des noyaux influencent également les structures des spectres moléculaires. En particulier, les moments de rotation (spins) des noyaux jouent un rôle très important. Si dans une molécule composée d'atomes identiques, les moments de rotation des noyaux sont égaux à zéro, une ligne sur deux de la bande de rotation disparaît. Cet effet est par exemple observé dans la molécule
Si les moments de rotation des noyaux sont non nuls, ils peuvent provoquer une alternance d'intensités dans la bande de rotation, des raies faibles alternant avec des raies fortes.)
Enfin, grâce à des méthodes de radiospectroscopie, il a été possible de détecter et de mesurer avec précision la structure hyperfine des spectres moléculaires associés au moment électrique quadripolaire des noyaux.
Le moment électrique quadripolaire résulte de la déviation de la forme nucléaire par rapport à la forme sphérique. Le noyau peut avoir la forme d'un ellipsoïde de révolution allongé ou aplati. Un tel ellipsoïde chargé ne peut plus être remplacé simplement par une charge ponctuelle placée au centre du noyau.

Riz. 291. Dispositif absorbant pour horloges « atomiques » : 1 - un guide d'onde rectangulaire de section transversale d'une longueur fermé de part et d'autre par des cloisons étanches aux gaz 7 et rempli d'ammoniac à basse pression ;
2 - diode à cristal qui crée des harmoniques de la tension haute fréquence qui lui est fournie ; 3 - diode à cristal de sortie ; 4 - générateur de tension haute fréquence modulée en fréquence ; 5 - pipeline vers la pompe à vide et le réservoir de gaz ammoniac ; 6 - sortie vers un amplificateur d'impulsions ; 7 - cloisons ; I - indicateur de courant à diode à cristal ; B - vacuomètre.
En plus de la force coulombienne, une force supplémentaire apparaît dans le champ nucléaire, inversement proportionnelle à la puissance quatrième de la distance et dépendant de l'angle avec la direction de l'axe de symétrie du noyau. L'apparition d'une force supplémentaire est associée à la présence d'un moment quadripolaire au niveau du noyau.
Pour la première fois, la présence d'un moment quadripolaire dans un noyau a été établie par spectroscopie conventionnelle en utilisant certains détails de la structure hyperfine des raies atomiques. Mais ces méthodes n'ont pas permis de déterminer avec précision l'ampleur du moment.
Dans la méthode radiospectroscopique, un guide d'ondes est rempli du gaz moléculaire étudié et l'absorption des ondes radio dans le gaz est mesurée. L'utilisation de klystrons pour générer des ondes radio permet d'obtenir des oscillations avec un degré élevé de monochromaticité, qui sont ensuite modulées. Le spectre d'absorption de l'ammoniac dans la région des ondes centimétriques a été étudié en détail et une structure hyperfine a été découverte dans ce spectre, qui s'explique par la présence d'une connexion entre le moment quadripolaire du noyau et le champ électrique de la molécule elle-même.
L'avantage fondamental de la radiospectroscopie est la faible énergie des photons correspondant aux radiofréquences. Grâce à cela, l’absorption des radiofréquences permet de détecter des transitions entre des niveaux d’énergie extrêmement proches des atomes et des molécules. En plus des effets nucléaires, la méthode de radiospectroscopie est très pratique pour déterminer les moments dipolaires électriques de la molécule entière par l'effet Stark des raies moléculaires dans des courants électriques faibles.
des champs. Ces dernières années, de très nombreux travaux ont été consacrés à la méthode radiospectroscopique pour étudier la structure d'une grande variété de molécules. L'absorption des ondes radio dans l'ammoniac a été utilisée pour construire des horloges « atomiques » ultra précises (Fig. 291).
La durée du jour astronomique augmente lentement et, en outre, fluctue dans les limites. Il est souhaitable de construire des horloges avec un rythme plus uniforme. Une horloge « atomique » est un générateur à quartz d’ondes radio dont la fréquence est contrôlée par l’absorption des ondes générées dans l’ammoniac. A une longueur d'onde de 1,25 cm, une résonance se produit avec la fréquence naturelle de la molécule d'ammoniac, ce qui correspond à une raie d'absorption très nette. Le moindre écart de la longueur d'onde du générateur par rapport à cette valeur perturbe la résonance et entraîne une forte augmentation de la transparence du gaz pour l'émission radio, qui est enregistrée par l'équipement approprié et active l'automatisme qui rétablit la fréquence du générateur. Les horloges « atomiques » se sont déjà déplacées de manière plus uniforme que la rotation de la Terre. On suppose qu’il sera possible d’atteindre une précision de l’ordre d’une fraction de journée.

spectres moléculaires, spectres d'émission et d'absorption optiques, ainsi que diffusion Raman, appartenant à des éléments libres ou faiblement connectés molécules. MS. ont une structure complexe. Typique M. s. - rayés, ils s'observent en émission et en absorption et en diffusion Raman sous la forme d'un ensemble de bandes plus ou moins étroites dans les domaines de l'ultraviolet, du visible et du proche infrarouge, qui se fragmentent avec le pouvoir de résolution suffisant des instruments spectraux utilisés en un ensemble de lignes rapprochées. La structure spécifique de M. s. est différent selon les molécules et, d’une manière générale, devient plus complexe à mesure que le nombre d’atomes dans la molécule augmente. Pour les molécules très complexes, les spectres visible et ultraviolet sont constitués de quelques larges bandes continues ; les spectres de ces molécules sont similaires les uns aux autres.
MS. surgir quand transitions quantiques entre niveaux d'énergie E' Et E''molécules selon le rapport
h m= E‘ - E‘’, (1)
Où h n - énergie émise absorbée photon fréquence n ( h -constante de Planck ). Avec diffusion Raman h n est égal à la différence entre les énergies des photons incidents et diffusés. MS. beaucoup plus complexe que les spectres atomiques linéaires, qui sont déterminés par la plus grande complexité des mouvements internes d'une molécule que de ceux des atomes. Parallèlement au mouvement des électrons par rapport à deux ou plusieurs noyaux dans les molécules, un mouvement vibrationnel des noyaux (ainsi que des électrons internes qui les entourent) se produit autour des positions d'équilibre et du mouvement de rotation de la molécule dans son ensemble. Ces trois types de mouvements – électronique, vibratoire et rotationnel – correspondent à trois types de niveaux d’énergie et trois types de spectres.
Selon la mécanique quantique, l’énergie de tous les types de mouvement dans une molécule ne peut prendre que certaines valeurs, c’est-à-dire qu’elle est quantifiée. Énergie totale d'une molécule E peut être approximativement représenté comme la somme des valeurs d'énergie quantifiées de trois types de son mouvement :
E = E email + E compter + E tourner (2)
Par ordre de grandeur
Où m est la masse de l'électron et la magnitude M a l'ordre de masse des noyaux atomiques dans une molécule, c'est-à-dire m/m~ 10 -3 -10 -5, donc :
E email >> E compter >> E tourner (4)
Généralement E el à propos de plusieurs ev(plusieurs centaines kJ/mole), E compter ~ 10 -2 -10 -1 eV, E rotation ~ 10 -5 -10 -3 ev.
Conformément à (4), le système de niveaux d'énergie d'une molécule est caractérisé par un ensemble de niveaux électroniques éloignés les uns des autres (valeurs différentes E el à E compter = E rotation = 0), niveaux vibratoires situés beaucoup plus près les uns des autres (valeurs différentes E compter à un moment donné E atterrir E rotation = 0) et des niveaux de rotation encore plus rapprochés (différentes valeurs E rotation à donnée E el et E compter).
Niveaux d'énergie électroniques ( E el dans (2) correspondent aux configurations d'équilibre de la molécule (dans le cas d'une molécule diatomique, caractérisée par la valeur d'équilibre r 0 distance internucléaire r. Chaque état électronique correspond à une certaine configuration d'équilibre et une certaine valeur E el; la valeur la plus basse correspond au niveau d'énergie de base.
L'ensemble des états électroniques d'une molécule est déterminé par les propriétés de sa couche électronique. En principe, les valeurs E el peut être calculé à l'aide de méthodes chimie quantique, cependant, ce problème ne peut être résolu qu’en utilisant des méthodes approximatives et pour des molécules relativement simples. Les informations les plus importantes sur les niveaux électroniques d'une molécule (l'emplacement des niveaux d'énergie électronique et leurs caractéristiques), déterminées par sa structure chimique, sont obtenues en étudiant sa structure moléculaire.
Une caractéristique très importante d'un niveau d'énergie électronique donné est la valeur Nombre quantique S, caractérisant la valeur absolue du moment de spin total de tous les électrons de la molécule. Les molécules chimiquement stables ont généralement un nombre pair d'électrons, et pour elles S= 0, 1, 2... (pour le niveau électronique principal, la valeur typique est S= 0, et pour les excités - S= 0 et S= 1). Niveaux avec S= 0 sont appelés singulet, avec S= 1 - triplet (puisque l'interaction dans la molécule conduit à leur division en c = 2 S+ 1 = 3 sous-niveaux) . AVEC radicaux libres ont, en règle générale, un nombre impair d'électrons, pour eux S= 1 / 2, 3 / 2, ... et la valeur est typique des niveaux principal et excité S= 1 / 2 (doublets se divisant en c = 2 sous-niveaux).
Pour les molécules dont la configuration d’équilibre présente une symétrie, les niveaux électroniques peuvent être classés davantage. Dans le cas de molécules diatomiques et triatomiques linéaires ayant un axe de symétrie (d'ordre infini) passant par les noyaux de tous les atomes , les niveaux électroniques sont caractérisés par les valeurs du nombre quantique l, qui détermine la valeur absolue de la projection du moment orbital total de tous les électrons sur l'axe de la molécule. Les niveaux avec l = 0, 1, 2, ... sont désignés respectivement S, P, D..., et la valeur de c est indiquée par l'index en haut à gauche (par exemple, 3 S, 2 p, ...). Pour les molécules avec un centre de symétrie, par exemple CO 2 et C 6 H 6 , tous les niveaux électroniques sont divisés en pairs et impairs, désignés par des indices g Et toi(selon que la fonction d'onde conserve son signe lorsqu'elle est inversée au centre de symétrie ou le change).
Niveaux d'énergie vibratoire (valeurs E count) peut être trouvé en quantifiant le mouvement oscillatoire, qui est approximativement considéré comme harmonique. Dans le cas le plus simple d'une molécule diatomique (un degré de liberté vibrationnel, correspondant à une modification de la distance internucléaire r) il est considéré comme harmonique oscillateur; sa quantification donne des niveaux d'énergie équidistants :
E compter = h n e (u +1/2), (5)
où n e est la fréquence fondamentale des vibrations harmoniques de la molécule, u est le nombre quantique vibrationnel, prenant les valeurs 0, 1, 2, ... Pour chaque état électronique d'une molécule polyatomique constituée de N atomes ( N³ 3) et ayant F degrés de liberté vibratoires ( F = 3N- 5 et F = 3N- 6 pour les molécules linéaires et non linéaires, respectivement), il s'avère F soi-disant vibrations normales avec des fréquences n je ( je = 1, 2, 3, ..., F) et un système complexe de niveaux vibratoires :
![]()
Où toi i = 0, 1, 2, ... sont les nombres quantiques vibrationnels correspondants. L’ensemble des fréquences de vibrations normales dans l’état électronique fondamental est une caractéristique très importante d’une molécule, en fonction de sa structure chimique. Tout ou partie des atomes de la molécule participent à une certaine vibration normale ; les atomes effectuent des vibrations harmoniques avec la même fréquence v je, mais avec des amplitudes différentes qui déterminent la forme de la vibration. Les vibrations normales sont divisées selon leur forme en étirement (dans lequel les longueurs des lignes de liaison changent) et en flexion (dans laquelle les angles entre les liaisons chimiques - angles de liaison - changent). Le nombre de fréquences de vibration différentes pour les molécules de faible symétrie (sans axes de symétrie d'ordre supérieur à 2) est égal à 2, et toutes les vibrations sont non dégénérées, tandis que pour les molécules plus symétriques il existe des vibrations doublement et triplement dégénérées (paires et triplets de vibrations qui correspondent en fréquence). Par exemple, dans une molécule triatomique non linéaire H 2 O F= 3 et trois vibrations non dégénérées sont possibles (deux étirements et une flexion). La molécule triatomique linéaire CO 2 la plus symétrique a F= 4 - deux vibrations non dégénérées (étirement) et une doublement dégénérée (déformation). Pour une molécule plate hautement symétrique C 6 H 6, il s'avère F= 30 - dix oscillations non dégénérées et 10 doublement dégénérées ; parmi celles-ci, 14 vibrations se produisent dans le plan de la molécule (8 en étirement et 6 en flexion) et 6 vibrations en flexion hors du plan - perpendiculaires à ce plan. La molécule tétraédrique CH 4 encore plus symétrique a f = 9 - une vibration non dégénérée (étirement), une doublement dégénérée (déformation) et deux triplement dégénérées (un étirement et une déformation).
Les niveaux d'énergie de rotation peuvent être trouvés en quantifiant le mouvement de rotation d'une molécule, en la traitant comme un solide avec certains moments d'inertie. Dans le cas le plus simple d'une molécule diatomique ou polyatomique linéaire, son énergie de rotation
![]()
Où je est le moment d'inertie de la molécule par rapport à un axe perpendiculaire à l'axe de la molécule, et M- moment de rotation. D'après les règles de quantification,
![]()
où est le nombre quantique de rotation J.= 0, 1, 2, ..., et donc pour E rotation reçue :
où la constante de rotation détermine l'échelle des distances entre les niveaux d'énergie, qui diminue avec l'augmentation des masses nucléaires et des distances internucléaires.
Différents types de M. s. surviennent lors de divers types de transitions entre les niveaux d’énergie des molécules. D'après (1) et (2)
D E = E‘ - E'' = D E el + D E compter + D E tourner, (8)
où change D E el, D E compter et D E la rotation des énergies électroniques, vibratoires et rotationnelles satisfont à la condition :
D E el >> D E compter >> D E tourner (9)
[les distances entre les niveaux sont du même ordre que les énergies elles-mêmes E el, E vieux et E rotation, satisfaisant la condition (4)].
En D E el ¹ 0, on obtient une microscopie électronique qui est observée dans les régions visible et ultraviolette (UV). Généralement en D E el ¹ 0 simultanément D E numéro 0 et D E rotation ¹ 0 ; différent D E compter pour un D donné E el correspondent à différentes bandes vibratoires, et différents D E rotation à D donné E el et d E compter - lignes de rotation individuelles dans lesquelles cette bande se divise ; une structure rayée caractéristique est obtenue.
![]()
Division en rotation de la bande vibratoire électronique 3805 de la molécule N 2
Un ensemble de rayures avec un D donné E el (correspondant à une transition purement électronique avec une fréquence v el = D E e-mail/ h) appelé système de bandes ; les bandes individuelles ont des intensités différentes en fonction des probabilités relatives de transitions, qui peuvent être calculées approximativement par des méthodes de mécanique quantique. Pour les molécules complexes, les bandes d'un système correspondant à une transition électronique donnée fusionnent généralement en une large bande continue ; plusieurs de ces bandes larges peuvent se chevaucher. Spectres électroniques discrets caractéristiques observés dans des solutions congelées de composés organiques . Les spectres électroniques (plus précisément électroniques-vibratoires-rotationnels) sont étudiés expérimentalement à l'aide de spectrographes et de spectromètres à optique en verre (pour la région visible) et en quartz (pour la région UV), dans lesquels des prismes ou des réseaux de diffraction sont utilisés pour décomposer la lumière en un spectre .
En D E el = 0, et D E compte ¹ 0, des résonances magnétiques oscillatoires sont obtenues, observées à courte distance (jusqu'à plusieurs µm) et au milieu (jusqu'à plusieurs dizaines µm) région infrarouge (IR), généralement en absorption, ainsi qu'en diffusion Raman de la lumière. En règle générale, simultanément D E rotation ¹ 0 et à un instant donné E Le résultat est une bande vibratoire qui se divise en lignes de rotation distinctes. Ils sont plus intenses dans les M. s oscillatoires. rayures correspondant à D tu = toi’ - toi'' = 1 (pour les molécules polyatomiques - D toi je = toi je' - toi je''= 1 en D toi k = toi k' - toi k'' = 0, où k¹je).
Pour les vibrations purement harmoniques, ces règles de sélection, interdire que d'autres transitions soient effectuées strictement ; pour les vibrations anharmoniques, apparaissent des bandes pour lesquelles D toi> 1 (harmoniques) ; leur intensité est généralement faible et diminue avec l'augmentation de D toi.
Les spectres vibrationnels (plus précisément vibration-rotationnel) sont étudiés expérimentalement dans le domaine IR en absorption à l'aide de spectromètres IR à prismes transparents au rayonnement IR ou à réseaux de diffraction, ainsi que de spectromètres de Fourier et en diffusion Raman à l'aide de spectrographes à grande ouverture (pour le région visible) par excitation laser.
En D E el = 0 et D E count = 0, on obtient des systèmes magnétiques purement rotationnels, constitués de lignes individuelles. Ils sont observés en absorption à distance (des centaines de µm)Région IR et notamment dans la région des micro-ondes, ainsi que dans les spectres Raman. Pour les molécules diatomiques et polyatomiques linéaires (ainsi que pour les molécules polyatomiques non linéaires assez symétriques), ces lignes sont équidistantes (sur l'échelle de fréquence) les unes des autres avec des intervalles Dn = 2 B en spectres d'absorption et Dn = 4 B dans les spectres Raman.
Les spectres de rotation purs sont étudiés en absorption dans la région IR lointaine à l'aide de spectromètres IR dotés de réseaux de diffraction spéciaux (échelettes) et de spectromètres de Fourier, dans la région des micro-ondes à l'aide de spectromètres micro-ondes (micro-ondes). , ainsi qu'en diffusion Raman à l'aide de spectrographes à haute ouverture.
Les méthodes de spectroscopie moléculaire, basées sur l'étude des micro-organismes, permettent de résoudre divers problèmes de chimie, de biologie et d'autres sciences (par exemple, détermination de la composition de produits pétroliers, de substances polymères, etc.). En chimie selon MS. étudier la structure des molécules. Electronique M. s. permettent d'obtenir des informations sur les coques électroniques des molécules, de déterminer les niveaux excités et leurs caractéristiques, et de trouver les énergies de dissociation des molécules (par convergence des niveaux vibrationnels d'une molécule vers les frontières de dissociation). Etude de l'oscillatoire M. s. permet de retrouver les fréquences de vibration caractéristiques correspondant à certains types de liaisons chimiques dans la molécule (par exemple, liaisons simples doubles et triples C-C, liaisons C-H, N-H, O-H pour les molécules organiques), divers groupes d'atomes (par exemple, CH 2 , CH 3 , NH 2), déterminer la structure spatiale des molécules, distinguer les isomères cis et trans. À cette fin, les spectres d’absorption infrarouge (IR) et Raman (RSS) sont utilisés. La méthode IR est devenue particulièrement répandue comme l'une des méthodes optiques les plus efficaces pour étudier la structure des molécules. Il fournit les informations les plus complètes en combinaison avec la méthode SKR. L'étude des résonances magnétiques rotationnelles, ainsi que de la structure rotationnelle des spectres électroniques et vibrationnels, permet d'utiliser des valeurs expérimentalement trouvées des moments d'inertie des molécules [qui sont obtenues à partir des valeurs des constantes de rotation, voir (7)] pour trouver avec une grande précision (pour des molécules plus simples, par exemple H 2 O) les paramètres de la configuration d'équilibre de la molécule - longueurs de liaison et angles de liaison. Pour augmenter le nombre de paramètres déterminés, les spectres de molécules isotopiques (notamment dans lesquelles l'hydrogène est remplacé par du deutérium) ayant les mêmes paramètres de configurations d'équilibre, mais des moments d'inertie différents, sont étudiés.
À titre d'exemple de l'utilisation de M. s. Pour déterminer la structure chimique des molécules, considérons la molécule de benzène C 6 H 6 . L'étude de sa M. s. confirme l'exactitude du modèle selon lequel la molécule est plate et que les 6 liaisons C-C du cycle benzénique sont équivalentes et forment un hexagone régulier avec un axe de symétrie du sixième ordre passant par le centre de symétrie de la molécule perpendiculaire à son avion. Electronique M. s. la bande d'absorption C 6 H 6 est constituée de plusieurs systèmes de bandes correspondant aux transitions du niveau pair du sol aux niveaux impairs excités, dont le premier est un triplet et les plus élevés sont des singulets. Le système de rayures est le plus intense dans la région de 1840 ( E 5 - E 1 = 7,0 ev), le système de bandes est le plus faible aux alentours de 3400 ( E 2 - E 1 = 3,8ev), correspondant à la transition singulet-triplet, ce qui est interdit par les règles de sélection approximatives du spin total. Les transitions correspondent à l'excitation de ce qu'on appelle. électrons p délocalisés dans tout le cycle benzénique ; Le diagramme de niveaux obtenu à partir des spectres moléculaires électroniques est en accord avec les calculs approximatifs de la mécanique quantique. Oscillatoire M. s. C 6 H 6 correspondent à la présence d'un centre de symétrie dans la molécule - les fréquences vibratoires qui apparaissent (actives) dans l'IRS sont absentes (inactives) dans le SRS et vice versa (ce qu'on appelle l'interdiction alternative). Sur les 20 vibrations normales de C 6 H 6 4 sont actives dans l'ICS et 7 sont actives dans le SCR, les 11 restantes sont inactives à la fois dans l'ICS et dans le SCR. Valeurs de fréquence mesurées (en cm -1): 673, 1038, 1486, 3080 (en ICS) et 607, 850, 992, 1178, 1596, 3047, 3062 (en TFR). Les fréquences 673 et 850 correspondent à des vibrations non planes, toutes les autres fréquences correspondent à des vibrations planes. Les vibrations planaires sont particulièrement caractéristiques de la fréquence 992 (correspondant à la vibration d'étirement des liaisons C-C, constituée d'une compression et d'un étirement périodiques du cycle benzénique), des fréquences 3062 et 3080 (correspondant aux vibrations d'étirement des liaisons C-H) et de la fréquence 607 (correspondant aux vibrations planaires). à la vibration de flexion du cycle benzénique). Les spectres vibrationnels observés de C 6 H 6 (et les spectres vibrationnels similaires de C 6 D 6) sont en très bon accord avec les calculs théoriques, qui ont permis de donner une interprétation complète de ces spectres et de retrouver les formes de toutes les vibrations normales.
De la même manière, vous pouvez utiliser M. s. déterminer la structure de diverses classes de molécules organiques et inorganiques, jusqu'à des molécules très complexes, telles que les molécules de polymères.
Conférence 12. Physique nucléaire. La structure du noyau atomique.
Cœur- c'est la partie centrale massive de l'atome autour de laquelle les électrons tournent sur des orbites quantiques. La masse du noyau est environ 4,10 3 fois supérieure à la masse de tous les électrons inclus dans l'atome. La taille du noyau est très petite (10 -12 -10 -13 cm), qui est environ 10 5 fois inférieur au diamètre de l'atome entier. La charge électrique est positive et en valeur absolue est égale à la somme des charges des électrons atomiques (puisque l'atome dans son ensemble est électriquement neutre).
Le noyau a été découvert par E. Rutherford (1911) lors d'expériences sur la diffusion des particules alpha lors de leur passage à travers la matière. Ayant découvert que les particules a sont dispersées sous de grands angles plus souvent que prévu, Rutherford a suggéré que la charge positive de l'atome est concentrée dans un petit noyau (avant cela, les idées de J. Thomson prévalaient, selon lesquelles la charge positive de l'atome était considéré comme uniformément réparti dans tout son volume). L'idée de Rutherford n'a pas été immédiatement acceptée par ses contemporains (le principal obstacle était la croyance en la chute inévitable des électrons atomiques sur le noyau en raison de la perte d'énergie due au rayonnement électromagnétique lors du déplacement en orbite autour du noyau). Les célèbres travaux de N. Bohr (1913), qui posèrent les bases de la théorie quantique de l’atome, jouèrent un rôle majeur dans sa reconnaissance. Bohr a postulé la stabilité des orbites comme principe initial de quantification du mouvement des électrons atomiques et en a ensuite dérivé les lois des spectres optiques linéaires qui expliquaient un vaste matériel empirique (série de Balmer, etc.). Un peu plus tard (à la fin de 1913), l'étudiant de Rutherford, G. Moseley, montra expérimentalement que le déplacement de la limite d'onde courte de la raie des spectres de rayons X des atomes lorsque le numéro atomique Z d'un élément dans le système périodique d'éléments change correspond à la théorie de Bohr, si l'on suppose que la charge électrique du noyau (en unités de charge électronique) est égale à Z. Cette découverte a complètement brisé la barrière de la méfiance : un nouvel objet physique - le noyau - s'est avéré être fermement connecté avec toute une série de phénomènes apparemment hétérogènes, qui ont désormais reçu une explication unifiée et physiquement transparente. Après les travaux de Moseley, l'existence du noyau atomique a finalement été établie en physique.
Composition du noyau. Au moment de la découverte du noyau, seules deux particules élémentaires étaient connues : le proton et l'électron. En conséquence, il a été jugé probable que le noyau en soit constitué. Cependant, à la fin des années 20. 20ième siècle L'hypothèse proton-électron s'est heurtée à une sérieuse difficulté, appelée « catastrophe de l'azote » : selon l'hypothèse proton-électron, le noyau d'azote devrait contenir 21 particules (14 protons et 7 électrons), dont chacune avait un spin de 1/2. . Le spin du noyau d'azote aurait dû être demi-entier, mais selon les données de mesure des spectres moléculaires optiques, le spin s'est avéré être égal à 1.
La composition du noyau a été clarifiée après la découverte par J. Chadwick (1932) neutron. La masse du neutron, comme l'ont montré les premières expériences de Chadwick, est proche de la masse du proton et le spin est égal à 1/2 (établi plus tard). L'idée selon laquelle le noyau est constitué de protons et de neutrons a été exprimée pour la première fois sous forme imprimée par D. D. Ivanenko (1932) et immédiatement après, elle a été développée par W. Heisenberg (1932). L'hypothèse concernant la composition proton-neutron du noyau a ensuite été pleinement confirmée expérimentalement. En physique nucléaire moderne, le proton (p) et le neutron (n) sont souvent combinés sous le nom commun de nucléon. Le nombre total de nucléons dans un noyau est appelé nombre de masse. UN, le nombre de protons est égal à la charge du noyau Z (en unités de charge électronique), le nombre de neutrons N = A-Z. U isotopes même Z, mais différent UN Et N, les noyaux ont les mêmes isobares UN et différents Z et N.
Dans le cadre de la découverte de nouvelles particules plus lourdes que les nucléons, ce qu'on appelle. isobares des nucléons, il s'est avéré qu'ils devraient également faire partie du noyau (les nucléons intranucléaires, entrant en collision les uns avec les autres, peuvent se transformer en isobares des nucléons). Dans le noyau le plus simple - deuton , constitués d'un proton et d'un neutron, les nucléons devraient rester sous forme d'isobares de nucléons environ 1 % du temps. Un certain nombre de phénomènes observés témoignent en faveur de l'existence de tels états isobares dans les noyaux. En plus des nucléons et des isobares des nucléons, dans les noyaux périodiquement pendant une courte période (10 -23 -10 -24 seconde) apparaître mésons , y compris le plus léger d'entre eux - les mésons p. L'interaction des nucléons se résume à de multiples actes d'émission d'un méson par l'un des nucléons et de son absorption par un autre. Émergeant, c'est-à-dire. Les courants d'échange de mésons affectent notamment les propriétés électromagnétiques des noyaux. La manifestation la plus distincte des courants d'échange de mésons a été trouvée dans la réaction de division du deuton par des électrons de haute énergie et des quanta g.
Interaction des nucléons. Les forces qui maintiennent les nucléons dans le noyau sont appelées nucléaire . Ce sont les interactions les plus fortes connues en physique. Les forces nucléaires agissant entre deux nucléons dans un noyau sont cent fois plus intenses que l’interaction électrostatique entre protons. Une propriété importante des forces nucléaires est leur. indépendance de l'état de charge des nucléons : les interactions nucléaires de deux protons, de deux neutrons, ou d'un neutron et d'un proton sont les mêmes si les états de mouvement relatif de ces paires de particules sont les mêmes. L'ampleur des forces nucléaires dépend de la distance entre les nucléons, de l'orientation mutuelle de leurs spins, de l'orientation des spins par rapport au moment cinétique orbital et du rayon vecteur tracé d'une particule à l'autre. Les forces nucléaires se caractérisent par une certaine amplitude d'action : le potentiel de ces forces diminue avec la distance r entre les particules plus rapidement que r-2, et les forces elles-mêmes sont plus rapides que r-3. De la nature physique des forces nucléaires, il s’ensuit qu’elles devraient diminuer de façon exponentielle avec la distance. Le rayon d'action des forces nucléaires est déterminé par ce qu'on appelle. Longueur d'onde Compton r 0 mésons échangés entre nucléons lors de l'interaction :
ici m, est la masse du méson, est la constante de Planck, Avec- vitesse de la lumière dans le vide. Les forces provoquées par l’échange de mésons p ont le plus grand rayon d’action. Pour eux r 0 = 1,41 F (1 f = 10 -13 cm). Les distances internucléaires dans les noyaux sont précisément de cet ordre de grandeur, mais les échanges de mésons plus lourds (mésons m, r, w, etc.) contribuent également aux forces nucléaires. La dépendance exacte des forces nucléaires entre deux nucléons sur la distance et la contribution des forces nucléaires dues à l'échange de mésons de types différents n'a pas été établie avec certitude. Dans les noyaux multinucléons, des forces sont possibles qui ne peuvent être réduites à l'interaction de seules paires de nucléons. Le rôle de ces soi-disant Les forces à plusieurs particules dans la structure des noyaux restent floues.
Tailles des noyaux dépendent du nombre de nucléons qu’ils contiennent. La densité moyenne du nombre p de nucléons dans un noyau (leur nombre par unité de volume) pour tous les noyaux multinucléons (A > 0) est presque la même. Cela signifie que le volume du noyau est proportionnel au nombre de nucléons UN, et sa taille linéaire ~UNE 1/3. Rayon de noyau effectif R. est déterminé par la relation :
R = une UNE 1/3 , (2)
où est la constante UN proche de Hz, mais en diffère et dépend des phénomènes physiques dans lesquels il est mesuré R.. Dans le cas du rayon de charge du noyau, mesuré par la diffusion des électrons sur les noyaux ou par la position des niveaux d'énergie m- mésoatomes : une = 1,12 F. Rayon effectif déterminé à partir des processus d'interaction hadrons (nucléons, mésons, particules a, etc.) avec des noyaux légèrement plus gros que la charge : à partir de 1,2 F jusqu'à 1,4 F.
La densité de la matière nucléaire est incroyablement élevée par rapport à la densité des substances ordinaires : elle est d'environ 10 14 g/cm 3. Au cœur, r est quasiment constant dans la partie centrale et décroît de façon exponentielle vers la périphérie. Pour une description approximative des données empiriques, la dépendance suivante de r sur la distance r du centre du noyau est parfois acceptée :
![]() .
.
Rayon de noyau effectif R.égal à R. 0 + b. La valeur b caractérise le flou de la limite du noyau ; elle est quasiment la même pour tous les noyaux (» 0,5 F). Le paramètre r 0 est la double densité à la « frontière » du noyau, déterminée à partir de la condition de normalisation (égalité de l'intégrale de volume de p au nombre de nucléons UN). De (2), il s'ensuit que les tailles des noyaux varient dans un ordre de grandeur de 10 à 13 cm jusqu'à 10h -12h cm pour les noyaux lourds (taille de l'atome ~ 10 -8 cm). Cependant, la formule (2) ne décrit l'augmentation des dimensions linéaires des noyaux avec une augmentation du nombre de nucléons qu'approximativement, avec une augmentation significative UN. Le changement de taille du noyau dans le cas de l'ajout d'un ou deux nucléons dépend des détails de la structure du noyau et peut être irrégulier. En particulier (comme le montrent les mesures du déplacement isotopique des niveaux d'énergie atomique), il arrive parfois que le rayon du noyau diminue même lorsque deux neutrons sont ajoutés.